REGISTRO DOI: 10.5281/zenodo.8397943
Aline Pimentel Caldeira1
Indra Liciane Nascimento De Freitas2
Isabella Rodrigues Vidal3
Ully Urzêda Basílio4
Adriana Mendonça Silva Alexandrino5
RESUMO
A síndrome de Mayer-Rokitansky-Küster-Hauser (MRKH) é uma doença congênita que causa aplasia do útero e parte da vagina em mulheres com características sexuais secundárias normais e cariótipo feminino. É diagnosticada pela amenorreia primária e afeta cerca de 1 em 5.000 nascidos vivos do sexo feminino. A síndrome MRKH pode ser de tipo I ou II, esta última associada a anomalias extragenitais. Sua causa exata é desconhecida, mas evidências sugerem fatores genéticos. O impacto psicossexual é significativo, exigindo aconselhamento. A ocorrência de leiomioma em remanescente uterino é rara, com desafios no diagnóstico e manejo, envolvendo exames de imagem e anatomopatologia. Este artigo tem como objetivo relatar o caso de uma paciente de 44 anos, com diagnóstico prévio de síndrome de Mayer Rokitansky-Küster-Hauser (MRKH), que apresentava dor pélvica crônica. Na investigação por imagem por ultrassonografia e por ressonância magnética, foram observadas duas massas pélvicas compatíveis com leiomiomas. Foi submetida a cirurgia eletiva, com ressecção das massas, em que o anatomopatológico diagnosticou leiomiomas. Foi realizada uma busca pelas palavras chaves “Rokitansky” e “Leiomioma” nas principais bases de dados Pubmed, Lilacs, Scielo e Google Scholar encontrados 38 relatos de casos semelhantes. Foi concluído que a presença de leiomiomatose em paciente com síndrome MRKH é rara, podendo apresentar quadros agudos ou crônicos, necessitando de exames de imagem para diagnóstico e que o manejo pode ser conservador ou cirúrgico por laparoscopia ou laparotomia, a depender do quadro clínico apresentado.
PALAVRAS-CHAVES: Mayer-Rokitansky-Küster-Hauser. Leiomiomatose.
ABSTRACT
Mayer-Rokitansky-Küster-Hauser syndrome (MRKH) is a congenital condition that causes uterine and vaginal aplasia in women with normal secondary sexual characteristics and female karyotype. It is diagnosed by primary amenorrhea and affects approximately 1 in 5,000 live female births. MRKH syndrome can be of type I or type II, the latter being associated with extragenital anomalies. Its exact cause is unknown, but evidence suggests genetic factors. The psychosocial impact is significant, requiring counseling. The occurrence of leiomyoma in residual uterine tissue is rare, with challenges in diagnosis and management, involving imaging and anatomopathology. This article aims to report the case of a 44-year-old patient with a previous diagnosis of Mayer-Rokitansky-Küster-Hauser syndrome (MRKH), who presented with pelvic pain two months ago. Imaging investigation by ultrasound and magnetic resonance imaging revealed two pelvic masses compatible with leiomyomas. She underwent elective surgery with resection of the masses, and the histopathological findings were consistent with leiomyomas. A search using the keywords “Rokitansky” and “Leiomyoma” in the main databases PubMed, Lilacs, Scielo, and Google Scholar yielded 38 similar case reports. It was concluded that the presence of leiomyomatosis in a patient with MRKH syndrome is rare and can present with acute or chronic symptoms, requiring imaging for diagnosis. Management can be conservative or surgical via laparoscopy or laparotomy, depending on the clinical presentation.
KEYWORDS: Mayer-Rokitansky-Küster-Hauser. Leiomyomatosis.
INTRODUÇÃO
A síndrome de Mayer-Rokitansky-Küster-Hauser (MRKH), também conhecida como aplasia Mülleriana, é uma doença congênita caracterizada por aplasia do útero e da parte superior da vagina em mulheres com características sexuais secundárias normais e um cariótipo feminino normal (46, XX)1.
A síndrome MRKH é classificada como tipo I (aplasia uterovaginal isolada) ou tipo II (associada a manifestações extragenitais). As anomalias extragenitais geralmente incluem malformações renais, esqueléticas, auditivas ou cardíacas. A etiologia da síndrome MRKH ainda permanece indefinida, no entanto, relatos crescentes de agrupamento familiar apontam para causas genéticas e o uso de várias técnicas genômicas permitiu a identificação de anomalias genéticas recorrentes promissoras em alguns pacientes.
A prevalência da síndrome está estimada em 1 em 5.000 nascidos vivos do sexo feminino2. A abordagem de diagnóstico e manejo, neste caso, é bastante desafiadora, necessitando de exames de imagem e muitas vezes conformados pelo exame anatomopatológico. A maioria das pacientes são assintomáticas, porém a amenorreia primária costuma ser o sinal clínico que leva a investigação e seu diagnóstico, ainda durante a adolescência. Em casos raros pode ocorrer leiomioma em remanescente uterino na síndrome MRKH. O impacto psicossexual de ter a síndrome MRKH não deve ser subestimado e os cuidados clínicos envolvem, acima de tudo, aconselhamento completo e apoio num diálogo cuidadoso com o paciente.
Esse relato de caso tem como objetivo exemplificar um caso raro de leiomiomatose em uma paciente com síndrome MRKH, além de fazer uma revisão bibliográfica dos relatos de casos semelhantes disponíveis nas principais bases de dados, proporcionando às comunidades médica e científica informações para um cuidado atencioso com essa população específica.
RELATO DE CASO
Paciente do sexo feminino, de 44 anos, branca, casada, com vida sexual ativa, nuligesta, dona de casa, residente em Planaltina – DF, teve diagnóstico de Síndrome de Rokitansky após investigação de amenorreia primária aos 18 anos. Perdeu acompanhamento de saúde na vida adulta. Referiu como comorbidades hipotireidismo em uso de levotiroxina 100mcg, esteatose hepática leve e dislipidemia sem tratamento.
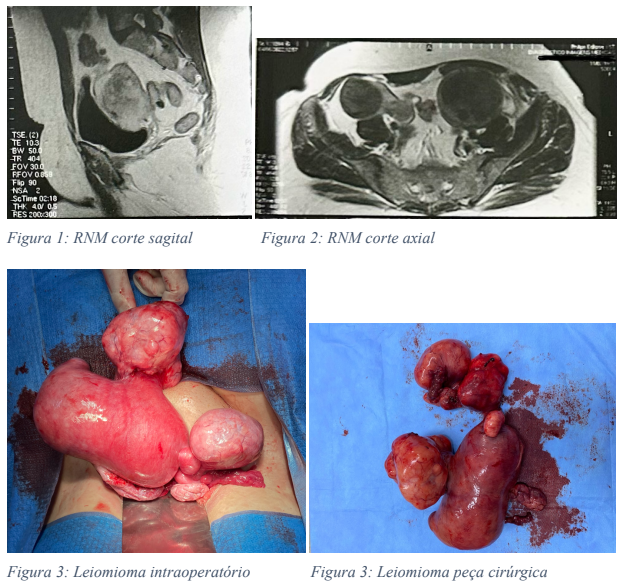
Referiu dor pélvica recorrente há dois meses, associada a perda de peso de 3kg em 2 meses, sem dieta ou exercício. Durante investigação em atenção primária, foi evidenciado em exame de imagem duas massas abdominais, sendo então encaminhada ao ambulatório de cirurgia ginecológica. Ao exame físico apresentava de baixa estatura, implantação capilar baixa, epicanto bilateral, aparente hipotelorismo, hipoplasia de face média, aparente micrognatia. Ao exame ginecológico apresentava vagina em fundo cego, funcional, de 4 cm, alcançada por atividade sexual, ausência de colo uterino. A ultrassonografia de abdome total apresentava esteatose hepática moderada, cisto cortical renal à esquerda, massa sólida heterogênea ocupando região pélvica e se estendendo para região anexial. Ao exame complementar de ressonância magnética de pelve apresentava ausência de útero, hiperplasia do canal vaginal, massas nas regiões anexiais com áreas nodulares de baixo sinal em T2 no seu interior e realce homogêneo pelo meio de contraste, cujas características são compatíveis com leiomiomas no tecido mulleriano remanescente. M1: 10 x 9 x 9 cm à esquerda e M2: 15 x 13 x 6 cm à direita. Divertículos no cólon sigmóide.
Foi realizada laparotomia exploradora, com visualização de massa abdominal compatível com útero com septo, apresentando à direita corno uterino miomatoso, com ovário e trompa à direita, além de foco de endometriose posterior, perto de ligamento uterossacro e à esquerda: massa compatível com corno uterino com ovário esquerdo, miomas subserosos, além de aderência de peritônio intestinal em mioma. Realizada retirada de massas compatíveis com útero e anexectomia bilateral. Paciente evoluiu com boa recuperação pós-cirúrgica, recebendo alta no segundo dia pós-operatório.
Na consulta pós-operatória, a paciente referiu fogachos principalmente noturnos, sem outras queixas álgicas. O exame anatomopatológico consiste no corpo de útero, deformado por nodulações, pesando 996,0 g e medindo 22,0 x 14,0 x 9,0 cm, sendo revestido por serosa espessada e congesta. Aos cortes, o miométrio apresenta múltiplos nódulos, bem delimitados, esbranquiçados, fasciculados, medindo o maior 9,0 x 7,0 cm, de localização submucosa, subserosa e intramural. Os ovários medem 3,5 x 3,0 cm e 5,0 x 2,5 cm, com superfície amarelada e lobulada. As tubas medem 6,0 x 1,0 cm e 9,0 x 1,0 cm, com lúmen virtual.
DISCUSSÃO
Noções embriológicas
O processo de diferenciação sexual começa na fecundação, onde o zigoto se torna XX ou XY3. Até a sexta semana do desenvolvimento fetal, os sistemas genitais são indiferenciados, compreendendo ductos mesonéfricos e paramesonéfricos. No embrião masculino, a produção do fator inibidor dos ductos de Müller (MIF) e testosterona pelas células de Sertoli e Leydig, respectivamente, leva à regressão dos ductos paramesonéfricos e diferenciação da genitália masculina. No sexo feminino, a ausência de MIF resulta na degeneração dos ductos mesonéfricos e desenvolvimento dos paramesonéfricos.
Os ductos de Müller contribuem para as estruturas reprodutivas e a gônada, formando as trompas de Falópio, útero e parte da vagina. Inicialmente separados por um septo, eles se fundem para formar o canal útero-vaginal. As tubas uterinas derivam de células distintas e não são afetadas por anormalidades dos ductos de Müller. Os ovários têm origem diferente e não são influenciados pelos ductos mesonéfricos e paramesonéfricos.
O sistema urinário compartilha origem com o sistema genital na fase inicial, podendo levar a anomalias renais como agenesia renal, ectopia renal cruzada, displasia cística e sistemas coletores duplos devido a diferenciações anormais nos ductos mesonéfricos e paramesonéfricos.
Diante do descrito, o momento que ocorre a alteração na embriogênese é importante patogênese das malformações uterinas:
• As aplasias ocorrem devido a ausência de formação ou parada de desenvolvimento dos ductos de Muller, uni ou bilateral, e ocorrem entre a sexta e a nona semana. Malformações urinárias podem acompanhar esses casos. Podem ser:
▪ Completas
▪ Unilaterais: útero bicorno
▪ Bilaterais: agenesia uterina, acompanhada de agenesia renal bilateral, que é incompatível a vida
▪ Incompletas
▪ Unilaterais: útero pseudounicorno
▪ Bilaterais: Síndrome Mayer-Rokitansky-Küster-Hauser
• Falhas na fusão dos ductos de Muller, entre 10ª e 12ª semanas, também podem acompanhar de malformações urinárias. Podem ser:
▪ Útero didelfo
▪ Útero bicorno bicervical
▪ Útero bicorno unicervical
• Problemas na reabsorção do septo intermulleriano, durante a 13ª e 17ª semanas, dão origem a úteros septados e as malformações urinárias geralmente estão ausentes.
Síndrome Mayer-Rokitansky-Kuster-Hauser
A síndrome MRKH é caracterizada pela ausência congênita do útero e de parte da vagina em mulheres com desenvolvimento normal de características sexuais secundárias e cariótipo 46, XX normal4. Pode ocorrer com outras malformações, como renais, esqueléticas, auditivas, cardíacas ou digitais. A forma isolada é denominada MRKH tipo I, enquanto a forma associada a outras malformações é chamada de MRKH tipo II (ou MURCS). A prevalência estimada é de 1 em 4.550 nascimentos femininos, geralmente de forma esporádica, embora casos familiares também ocorram. O padrão de herança parece ser autossômico dominante com graus variáveis de expressão. O MRKH tipo II é mais comum que o tipo I.
As pacientes geralmente não apresentam sintomas até a amenorreia primária. Seu fenótipo é tipicamente feminino, com características sexuais secundárias normais. A avaliação revela uma vagina encurtada, semelhante a uma covinha, com profundidade variável (2 a 7 cm).
O diagnóstico e classificação da síndrome MRKH requerem estudo anatômico. O tipo I envolve a completa ausência de útero, com dois cornos rudimentares conectados por uma dobra peritoneal e trompas de Falópio normais. O tipo II apresenta hipoplasia uterina simétrica ou assimétrica, frequentemente com um corno ausente ou diferença de tamanho entre os rudimentos dos cornos, além de malformações tubárias como hipoplasia ou aplasia de um ou ambos os tubos.
Leiomiomatose e MRKH
O leiomioma uterino é um tumor benigno, formado por fibras musculares lisas, entrelaçadas por tecido conectivo2. A idade é o principal fator de risco, com maior incidência entre 35 e 50 anos. Mutações somáticas no miométrio resultam na perda da capacidade de regular o crescimento dessas células, culminando com um novo fenótipo. Esse processo gradual e progressivo de transformação celular leva à formação de um novo tipo de crescimento. Tratam-se de tumores hormônio dependentes, nos quais estradiol e progesterona promovem seu crescimento durante a menacme. Em contrapartida, a diminuição dos níveis circulantes promove sua regressão. Esses dados corroboram com os relatos revisados, pois as pacientes com a síndrome MRKH podem possuir células miometriais nos cornos uterinos rudimentares, além de ter a capacidade hormonal sem alterações, já que possuem o eixo hipotálamo-hipófise-ovário normal.
REVISÃO DE LITERATURA
Foram encontrados 38 artigos com os temas “Rokitansky”e “Leiomioma” nas bases de dados Pubmed, Lilacs, Scielo e Google Scholar5-41. As pacientes foram diagnosticadas com massas pélvicas, em sua maioria realizadas cirurgias que corroboram com o diagnóstico de leiomiomatose nos cornos uterinos rudimentares. A média de idade das pacientes foi de 41 anos, sendo a máxima 70 anos e a mínima 16 anos. Os sintomas mais frequentes foram dor pélvica crônica, massa abdominal palpável, sintomas urinários como incontinência e retenção, distensão abdominal, algumas assintomáticas com diagnóstico incidental e 4 casos de abdome agudo com torção da massa pélvica.
Os exames de imagens que foram utilizados para diagnósticos das massas foram a ultrassonografia (USG) e a ressonância magnética (RNM), que é superior a à tomografia computadorizada (TC) ao mostrar detalhadamente as estruturas müllerianas (restos uterinos ou agenesia completa), incluindo a presença de endométrio em remanescentes uterinos. A RNM também mostra os ovários e malformações extragenitais e tem alta concordância entre avaliadores com a laparoscopia. No caso da paciente relatada, foi realizado USG para investigação de dor pélvica, complementado com a RNM, que delimitou as massas pélvicas.
Como condutas 18 casos encontrados na literatura foram para laparotomia, 15 casos por videolaparoscopia (VLP), 2 casos começaram com VLP e foram convertidos para cirurgia aberta, 1 caso foi descrito cirurgia endoscópica transluminal de orifício natural transvaginal (NOTAS), 1 foi optado por tratamento conservador, 1 a paciente recusou o tratamento. Os tamanhos dos miomas variaram de pequenos com 2 cm a grandes com 25 cm de diâmetro. Em todos foram usados exames de imagem, como ultrassonografia, ressonância magnética e tomografia computadorizada, em alguns casos sendo necessário os três para elucidação das massas. Alguns casos foram associados a patologias ovarianas, como endometrioma e tumor de ovário.
Além disso, existe a possibilidade de complicação do quadro, evoluindo para abdome agudo, em caso de torção das massas miomatosas, como descrito em 9 artigos. Essas pacientes apresentaram dor aguda e sintomas sistêmicos como náuseas, vômitos, inapetência, distensão abdominal, se configurando um quadro possível de emergência ginecológica. Em alguns casos, houve torção de ovário em associação ao quadro. Sendo necessário conduta cirúrgica. A paciente relatada teve toda sua propedêutica conduzida de modo eletivo.
A indicação do tratamento do leiomioma é individualizada e leva em consideração inúmeros fatores, como: sintomas, idade da paciente, número, tamanho e localização dos miomas, expectativa em relação ao futuro reprodutivo e desejo de preservar o útero, tratamentos prévios, além da coexistência de outras doenças. Nas pacientes com MRKH, a infertilidade é um fator que contribui para a possibilidade de tratamento cirúrgico. Dos 38 relatos, apenas dois não foram realizados cirurgia, sendo um deles por motivos da paciente estar assintomática e pelo tamanho do mioma de 6 cm e o outros por ter sido um achado acidental de uma paciente internada por outros motivos clínicos, que recusou o tratamento cirúrgico. Dentre os outros em que foram realizadas cirurgias, a escolha entre VLP e laparotomia depende do serviço hospitalar, do tamanho do mioma, da expertise do médico cirurgião. No caso em questão, foi optado por laparotomia pelo tamanho das massas abdominais e por ter maior possibilidade da cirurgia aberta no serviço, em comparação a VLP.
Tabela 1: comparação entre artigos de relato de caso.

CONCLUSÃO
Em mulheres que sofrem da Síndrome de Mayer-Rokitansky-Küster-Hauser e experimentam dor pélvica que não melhora com tratamento ambulatorial para a dor ou distensão e massa abdominal, é importante sempre considerar a possibilidade de uma complicação ginecológica como um dos diagnósticos potenciais. Recomenda-se a utilização de ultrassonografia como primeira abordagem, podendo ser complementada com RNM ou TC para uma avaliação anatômica mais detalhada e uma melhor preparação para a cirurgia. No entanto, devido à raridade e à complexidade anatômica dessas condições, o diagnóstico definitivo só pode ser estabelecido por meio de procedimento cirúrgico e análise histológica.
Por fim, com base nessa revisão, destacamos que a conduta sobre os leiomiomas pode ser semelhante a das pacientes sem a síndrome MRKH, ou seja, conservadora ou cirúrgica. O método de cirurgia escolhido depende do tamanho do tumor, da expertise do cirurgião, dos recursos oferecidos pelo centro hospitalar. Além disso, é importante ratificar a evolução para torção dessas massas pélvicas, configurando um abdome agudo. Sendo assim, é possível que pacientes submetidas a procedimentos cirúrgicos na região pélvica se beneficiem da remoção preventiva de qualquer tecido uterino residual por meio de cirurgia minimamente invasiva, reduzindo o desconforto para as pacientes.
REFERÊNCIAS
1. BAGNOLI, V.R. et al. Conduta frente às malformações genitais uterinas: revisão baseada em evidências. Femina, v. 38, n. 4, abr. 2010.
2. FERNANDES, C.E; SÁ, M.F.S de (Editores); SILVA FILHO, A.L da (Coordenação) et al. Tratado de ginecologia Febrasgo. 1. ed. Rio de Janeiro: Elsevier, 2019.
3. MORCEL, K.; CAMBORIEUX, L.; GUERRIER, D. Mayer-Rokitansky-Küster Hauser (MRKH) syndrome. Orphanet J Rare Dis, v. 2, 13, 2007. doi: 10.1186/1750- 1172-2-13.
4. HERLIN, M.K.; PETERSEN, M.B.; BRÄNNSTRÖM, M. Mayer-Rokitansky Küster-Hauser (MRKH) syndrome: a comprehensive update. Orphanet J Rare Dis, v. 15, 214, 2020. doi: 10.1186/s13023-020-01491-9.
5. RHEE, C.S.; KIM, J.S.; WOO, S.K.; SUH, S.J. MRI of round ligament leiomyoma associated with Mayer-Rokitansky-Kuster-Hauser syndrome. Abdom Imaging, v. 24, n. 2, p. 202-204, 1999. doi: 10.1007/s002619900478.
6. TSIN, D.A.; WATERS, T.K.; GRANATO, R.C. Laparoscopic myomectomy in a patient with Mayer-Rokitansky-Kuster-Hauser syndrome. J Am Assoc Gynecol Laparosc, v. 7, n. 3, p. 411-413, 2000. doi: 10.1016/s1074-3804(05)60488-4.
7. JAIN, N.; KRIPLANI, I.; SHARMA, S.; HANUMANTAIYA, S.; KRIPLANI, A. Urinary retention unveiling deeply embedded multiple leiomyomas in women with Mayer-Rokitansky-Kuster-Hauser syndrome and its successful laparoscopic management: a case-report and literature review. J Surg Case Rep, 2022, rjac291. doi: 10.1093/jscr/rjac291.
8. GALAJDOVA, L.; VERBEKEN, K.; DHONT, M. Recurrent multiple leiomyomata in a patient with Mayer-Rokitansky-Küster-Hauser syndrome. J Obstet Gynaecol, v. 23, n. 4, p. 448-449, 2003. doi: 10.1080/0144361031000122714.
9. EDMONDS, D.K. Multiple fibroids in a postmenopausal woman with Mayer Rokitansky Kuster Hauser syndrome. J Pediatr Adolesc Gynecol, v. 16, n. 2, p. 65- 66, 2003. doi: 10.1016/s1083-3188(03)00013-5.
10. DEKAR, D.; GUPTA, N.; DADHWAL, V.; MITTAL, S. Development of leiomyomas on the uterine remnants of two women with Mayer-Rokitansky Küster-Hauser syndrome. Fertil Steril, v. 81, n. 5, p. 1385-1387, 2004. doi: 10.1016/j.fertnstert.2003.09.067.
11. ITO, Y.; KOMORI, S.; HORIUCHI, I.; KINUTA, T.; HORI, M.; WADA, C.; TSUBAMOTO, H.; TANAKA, H.; K. Koyama. Solid pelvic tumor in a woman with Mayer-Rokitansky-Küstner-Hauser syndrome. Arch Gynecol Obstet, v. 274, p. 252–254, 2006.
12. PAPA, G.; ANDREOTTI, M.; GIANNUBILO, S.R.; CESARI, R.; CERÉ, I.; TRANQUILLI, A.L. Case report and surgical solution for a voluminous uterine leiomyoma in a woman with complicated Mayer-Rokitansky-Küster-Hauser syndrome. Fertil Steril, v. 90, n. 5, p. 2014.e5-6, 2008. doi: 10.1016/j.fertnstert.2008.04.061.
13. LANOWSKA, M.; FAVERO, G.; SCHNEIDER, A.; KÖHLER, C. Laparoscopy for differential diagnosis of a pelvic mass in a patient with Mayer-Rokitansky Küster-Hauser (MRKH) syndrome. Fertil Steril, v. 91, n. 3, p. 931.e17-8, 2009. doi: 10.1016/j.fertnstert.2008.09.057.
14. FUKUDA, J.; KUMAZAWA, Y.; FUJIMOTO, T.; TANAKA, T. Mayer Rokitansky-Kustner Hauser syndrome complicated by either uterine leiomyoma or ovarian tumor. J Obstet Gynaecol Res, v. 36, n. 1, p. 191-194, 2010. doi: 10.1111/j.1447-0756.2009.01116.x.
15. DEKA, D.; GUPTA, N.; DADHWAL, V.; MITTAL, S. Laparoscopic evaluation of pelvic pain with Surgicel vaginoplasty in a woman with Mayer Rokitansky Kuster Hauser syndrome. Eur J Obstet Gynecol Reprod Biol, v. 154, n. 1, p. 115- 116, 2011. doi: 10.1016/j.ejogrb.2010.07.042.
16. FLETCHER, H.M.; CAMPBELL-SIMPSON, K.; WALCOTT, D.; HARRIOTT, J. Müllerian remnant leiomyomas in women with Mayer-Rokitansky-Küster Hauser syndrome. Obstet Gynecol, v. 119, n. 2 Pt 2, p. 483-485, 2012. doi: 10.1097/AOG.0b013e318242a9b5.
17. VAN TROOST, M.H.W.; GEOMINI, P.M.A.J.; BONGERS, M.Y. Laparoscopic removal of a large uterine leiomyoma in a patient with Mayer–Rokitansky– Küster–Hauser syndrome: a case report. Gynecol Surg, v. 9, p. 445–447, 2012. doi: 10.1007/s10397-012-0747-3.
18. RAWAT, K.S.; BUXI, T.; YADAV, A.; GHUMAN, S.S.; DHAWAN, S. Large leiomyoma in a woman with Mayer-Rokitansky-Kuster-Hauser syndrome. J Radiol Case Rep, v. 7, n. 3, p. 39-46, 2013. doi: 10.3941/jrcr.v7i3.1267.
19. PAEZ-LOPEZ, Guillermo et al. Manejo laparoscópico de miomatosis uterina en pacientes con Síndrome de Rokitansky: Reporte de un caso y revisión de la literatura. Rev Colomb Obstet Ginecol, v. 64, n. 4, p. 469-474, Dec. 2013.
20. SALMAN, S.; BOZKURT, M.; YUMRU, A.E.; BOZYIGIT, A.; KAVSI, B. et al. Laparoscopic Management of Leiomyoma Developing from Rudimentary Horn in Mayer- Rokitansky-Küster-Hauser Syndrome. J Androl Gynaecol, v. 1, n. 2, 2013.
21. KUNDU, K.; COHEN, A.W.; GOLDBERG, J. Acute torsion of uterine remnant leiomyoma with Mayer-Rokitansky-Küster-Hauser syndrome. Fertil Steril, v. 102, n. 2, p. 607-609, 2014. doi: 10.1016/j.fertnstert.2014.04.034.
22. BHUYAR, S.A. Rare case of leiomyoma in Mayer-Rokitansky-Kuster-Hauser syndrome. Int J Reprod Contracept Obstet Gynecol, v. 3, p. 488-490, 2014.
23. VIDYASHREE, P.G.; MURALIDHAR, P.V.; JAYARAM, N.; LATHA, K. Mayer Rokitansky-Kuster-Hauser syndrome with multiple leiomyomas. Int J Gynaecol Obstet, v. 128, n. 3, p. 270-271, 2015. doi: 10.1016/j.ijgo.2014.09.012.
24. HASEGAWA, A.; IGARASHI, H.; OHTA, T.; KURACHI, H.; TAKAHASHI, K. Three-dimensional computed tomography of pelvic masses in Mayer-Rokitansky Küster-Hauser syndrome. Obstet Gynecol, v. 125, n. 2, p. 393-396, 2015. doi: 10.1097/AOG.0000000000000646.
25. KULKARNI, M.M.; DESHMUKH, S.D.; HOL, K.; NENE, N. A rare case of Mayer Rokitansky-Kuster-Hauser syndrome with multiple leiomyomas in hypoplastic uterus. J Hum Reprod Sci, v. 8, n. 4, p. 242-244, 2015. doi: 10.4103/0974- 1208.170418.
26. NARAYANAN, R.; MARIAPPAN, S.; PAULRAJ, S.; SHANKAR, B. Imaging of leiomyomas arising from Müllerian remnants in a case of Mayer-Rokitansky Küster-Hauser syndrome. BMJ Case Rep, 2015, bcr2015210737. doi: 10.1136/bcr 2015-210737.
27. HOO, P.S.; NORHASLINDA, A.R.; REZA, J.N. Rare Case of Leiomyoma and Adenomyosis in Mayer-Rokitansky-Kuster-Hauser Syndrome. Case Rep Obstet Gynecol, 2016, 3725043. doi: 10.1155/2016/3725043.
28. SALEM WEHBE, G.; BITAR, R.; ZREIK, T.; SAMAHA, M.; WALTER, C.; SLEIMAN, Z. Intra-peritoneal leiomyoma of the round ligament in a patient with Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Facts Views Vis Obgyn, v. 8, n. 4, p. 233-235, 2016.
29. PARK, J.W.; HWANG, D.W.; JANG, J.A.; CHOI, Y.J.; CHUN, K.C.; KIM, Y.A. Leiomyoma mimicking a pelvic tumour in Mayer-Rokitansky-Küster-Hauser syndrome: A case report. J Obstet Gynaecol, v. 36, n. 3, p. 279-280, 2016. doi: 10.3109/01443615.2015.1060204.
30. SHARMA, R.; GULERIA, K.; SUNEJA, A.; BHASKARAN, S.; TANVEER, N. Giant leiomyoma with extensive myxoid degeneration in Mayer-Rokitansky Küster-Hauser syndrome. Int J Gynaecol Obstet, v. 138, n. 1, p. 125-127, 2017. doi: 10.1002/ijgo.12162.
31. YI, S.; XU, B.; ZENG, F.; XUE, M. Acute Torsion of Paraovarian Cyst and Ipsilateral Uterine Remnant Leiomyoma in a Patient with Mayer-Rokitansky Küster-Hauser Syndrome. J Minim Invasive Gynecol, v. 24, n. 1, p. 6-7, 2017. doi: 10.1016/j.jmig.2016.04.007.
32. AMARATUNGA, T.; KIRKPATRICK, I.; YAN, Y.; KARLICKI, F. Ectopic Pelvic Fibroid in a Woman With Uterine Agenesis and Mayer-Rokitansky-Küster Hauser Syndrome. Ultrasound Q, v. 33, n. 3, p. 237-241, 2017. doi: 10.1097/RUQ.0000000000000284.
33. BLONTZOS, N.; IAVAZZO, C.; VORGIAS, G.; KALINOGLU, N. Leiomyoma development in Mayer-Rokitansky-Küster-Hauser syndrome: a case report and a narrative review of the literature. Obstet Gynecol Sci, v. 62, n. 4, p. 294-297, 2019. doi: 10.5468/ogs.2019.62.4.294.
34. JOKIMAA, V.; VIRTANEN, J.; KUJARI, H.; ALA-NISSILÄ, S.; RANTANEN, V. A Mayer-Rokitansky-Küster-Hauser patient with leiomyoma and dysplasia of neovagina: a case report. BMC Womens Health, v. 20, n. 1, p. 157, 2020. doi: 10.1186/s12905-020-01026-1.
35. ALBAHLOL, I.A.; ELSHAMY, M.; EL-HADY, H.A.F.; ABD-ELWAHAB, E.M. Leiomyomas in a case of Mayer-Rokitansky-Kuster-Hauser syndrome: Case report. Eur J Obstet Gynecol Reprod Biol, v. 244, p. 199-200, 2020. doi: 10.1016/j.ejogrb.2019.10.043.
36. IBIDAPO-OBE O, OKUDO J, FILANI O. Incidental Finding of Leiomyoma in Mayer-Rokitansky-Kuster-Hauser Syndrome. J Investig Med High Impact Case Rep, v. 9, 23247096211014690, Jan-Dec 2021. doi: 10.1177/23247096211014690.
37. QIU, S.; XIE, Y.; ZOU, Y.; WANG, F. Giant hysteromyoma after vaginoplasty in a woman with Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: case report and review of the literature. J Int Med Res, v. 49, n. 12, 2021. doi: 10.1177/03000605211066394.
38. YANG, C.Y.; WANG, Y.T.; HUANG, C.C.; YA-WEN HSUEH, E.; LIN, W.C. Mayer-Rokitansky-Küster-Hauser syndrome with leiomyomas in a rudimentary uterus treated with transvaginal NOTES. Taiwan J Obstet Gynecol, v. 60, n. 5, p. 971-972, 2021. doi: 10.1016/j.tjog.2021.07.044.
39. HARZIF, A.K.; AMBALAGEN, S.; CHARILDA, F.E.; MUTIA, H.D. A rare case of multiple leiomyomas on rudimentary uterus in a woman with Mayer Rokitansky Kuster Hauser syndrome: A challenging diagnosis and laparoscopic approach. Int J Surg Case Rep, v. 81, 105711, Apr 2021. doi: 10.1016/j.ijscr.2021.105711.
40. ROMANO, F. et al. The Rare, Unexpected Condition of a Large Uterine Leiomyoma During Laparoscopic Utero-vaginal Anastomosis for Müllerian Agenesis (MRKH Syndrome). J Minim Invasive Gynecol, v. 29, n. 4, p. 828-829, May 2022. doi: 10.1016/j.jmig.2021.11.007.
1Médica Residente em Ginecologia e Obstetrícia no Hospital Regional de Sobradinho. Contato para Correspondência: alinepimentelcaldeira@gmail.com
2Médica Residente em Ginecologia e Obstetrícia no Hospital Regional de Sobradinho
3Médica Residente em Ginecologia e Obstetrícia no Hospital Regional de Sobradinho
4Médica Residente em Ginecologia e Obstetrícia no Hospital Regional de Sobradinho
5Médica Especialista em Ginecologia e Obstetrícia no Hospital Regional de Sobradinho