WILSON DISEASE: IMPACTS OF LATE DIAGNOSIS AND THERAPEUTIC STRATEGIES IN CASES WITH PROGRESSIVE DISEASE ADVANCEMENT.
REGISTRO DOI: 10.69849/revistaft/dt10202511132237
Eduarda Aparecida Garcia1
Aline Nardelli Grossklas1
RESUMO
A Doença de Wilson (DW) é um distúrbio autossômico recessivo raro que resulta de mutações no gene ATP7B, levando ao acúmulo de cobre em órgãos vitais como fígado, cérebro e olhos. Este estudo tem como objetivo discutir os impactos clínicos e terapêuticos do diagnóstico tardio da DW, analisando suas consequências no prognóstico e reforçando a importância da detecção precoce. Foi realizada uma revisão sistemática da literatura, conforme as diretrizes PRISMA 2020, com busca em bases de dados como PubMed, SciELO, LILACS e Google Acadêmico, incluindo publicações entre 2007 e 2024. Foram selecionados 18 artigos que atenderam aos critérios de inclusão, abordando aspectos clínicos, diagnósticos e terapêuticos da DW. Os resultados evidenciam que o atraso no diagnóstico compromete a eficácia terapêutica e aumenta a morbimortalidade, frequentemente levando à necessidade de transplante hepático. Conclui-se que a capacitação profissional e a implementação de protocolos diagnósticos padronizados são essenciais para reduzir complicações e melhorar o prognóstico dos pacientes com DW.
Palavras-chave: Doença de Wilson. Diagnóstico precoce. Alterações hepáticas. Gene ATP7B. Transplante hepático.
ABSTRACT
Wilson’s Disease (WD) is a rare autosomal recessive disorder caused by mutations in the ATP7B gene, leading to copper accumulation in vital organs such as the liver, brain, and eyes. This study aimed to discuss the clinical and therapeutic impacts of late diagnosis of WD, analyzing its prognostic consequences and emphasizing the importance of early detection. A systematic literature review was conducted following PRISMA 2020 guidelines, searching databases such as PubMed, SciELO, LILACS, and Google Scholar for publications between 2007 and 2024. Eighteen studies were included, addressing clinical, diagnostic, and therapeutic aspects of WD. Results indicate that delayed diagnosis compromises treatment efficacy and increases morbidity and mortality, often leading to liver transplantation. It is concluded that professional training and the implementation of standardized diagnostic protocols are essential to reduce complications and improve patient outcomes.
Keywords: Wilson’s Disease. Early diagnosis. Hepatic alterations. ATP7B gene. Liver transplantation.
1. INTRODUÇÃO
A Doença de Wilson (DW) é um distúrbio autossômico recessivo raro, causado por mutação no gene ATP7B, localizado no cromossomo 13, tal qual é passada de ambos os pais para os filhos. Essa mutação compromete a excreção biliar do cobre, levando a um acúmulo no organismo, principalmente no fígado, cérebro e olhos, resultando em manifestações clínicas hepáticas, neurológicas, psiquiátricas e oftalmológicas (CARTAXO, 2021).
Descrita pela primeira vez por Samuel Alexander Kinnier Wilson, em 1912, a doença tem sido estudada com importantes contribuições de pesquisadores como Kayser (1902) e Fleischer (1903), que identificaram os anéis pigmentares oculares, quando a patologia afeta a área oftalmológica, formando anéis na coloração marrom-dourado ao redor da íris dos olhos (PFEIFFER, 2007). Outros estudos mostraram a relação entre a doença e o metabolismo anormal do cobre, sendo a deficiência de ceruloplasmina, uma proteína responsável para o transporte de cobre no organismo, identificada como um marcador diagnóstico relevante (LINDER, 2018).
A epidemiologia da DW revela uma prevalência aproximada de 30 casos por milhão, com incidência de 1:30.000 a 1:40.000 nascimentos (PFEIFFER, 2007). A apresentação clínica é variável e pode ocorrer em qualquer faixa etária, sendo mais frequente entre 5 e 35 anos e sem predominância de sexo. Em crianças, as manifestações hepáticas são mais características do que outros sintomas, enquanto as alterações neurológicas e psiquiátricas são mais comuns em adultos jovens, desde a rigidez muscular até a depressão (CARTAXO, 2021).
O diagnóstico da DW é complexo, pois não há um exame único que a defina. Ele é baseado em critérios clínicos, laboratoriais e genéticos, incluindo a dosagem de ceruloplasmina, urina de 24 horas e cobre sérico livre. O tratamento deve ser realizado precocemente para evitar a progressão da doença e minimizar os seus danos. A terapia envolve o uso contínuo de quelantes de cobre, como a penicilamina e acetato de zinco, podendo, em casos graves, ser necessário o transplante hepático (PFEIFFER, 2007).
Dessa forma, este estudo busca discutir o impacto do diagnóstico tardio, suas consequências clínicas e terapêuticas, e a importância da capacitação dos profissionais de saúde na suspeição e manejo da DW.
2. FUNDAMENTAÇÃO TEÓRICA OU REVISÃO DA LITERATURA
Contexto Histórico
A Doença de Wilson foi descrita pela primeira vez de maneira sistemática por Samuel Alexander Kinnier Wilson, em 1912, em sua tese de doutorado publicada na revista Brain. Neste estudo, Wilson foi pioneiro ao perceber que alguns pacientes apresentavam uma combinação de problemas neurológicos progressivos, como tremores e dificuldades de movimento, junto com doenças hepáticas, como cirrose. Ele foi o primeiro a sugerir que essas condições estavam relacionadas a um distúrbio no metabolismo do cobre, um elemento essencial para o corpo, mas que, em excesso, se acumula em diversos órgãos, como o fígado e o cérebro, causando danos graves (DOOLEY, 2024).
A importância do trabalho de Wilson foi enorme, pois até então as doenças hepáticas e neurológicas eram frequentemente tratadas como condições separadas. Sua pesquisa ajudou a compreender como essas duas áreas poderiam estar interligadas por uma única doença. A partir dessa descrição inicial, os médicos passaram a investigar mais profundamente essa relação, o que, com o tempo, levou ao desenvolvimento de tratamentos e exames mais eficazes (PFEIFFER, 2007).
Em 1913, Rumpel demonstrou que havia aumento do teor de cobre em tecidos de pacientes acometidos, sendo um dos primeiros a sugerir que um defeito no metabolismo do cobre estava implicado a fisiopatologia da doença (ROBERTS; CHILSKY, 2008).
Epidemiologia
A DW é considerada rara, com prevalência estimada entre 1:30.000 e 1:40.000 nascidos vivos, afetando igualmente ambos os sexos. Estudos recentes sugerem prevalência maior em função do subdiagnóstico, especialmente em países em desenvolvimento. A manifestação clínica geralmente ocorre entre 5 e 35 anos, embora casos em crianças e idosos também tenham sido descritos (CZŁONKOWSKA et al., 2018).
Genética e Fisiopatologia
A Doença de Wilson é causada por mutações no gene ATP7B, localizado no cromossomo 13, que codifica uma proteína fundamental para o transporte de cobre para dentro da bile e para a incorporação do cobre na ceruloplasmina (FERENCI et al., 2019). Aproximadamente 300 mutações diferentes foram descritas, com destaque para a mutação H1069Q em populações da Europa Central e do Norte. De acordo com Ferenci et al. (2019), “a identificação precoce das mutações específicas permite estabelecer estratégias de rastreamento familiar mais eficazes”.
Isso significa que ao identificar as mutações genéticas de uma pessoa, é possível realizar um acompanhamento mais cuidadoso de familiares que podem estar em risco, permitindo o diagnóstico precoce e evitando o agravamento da doença. Esse tipo de rastreamento é fundamental, já que a Doença de Wilson pode se manifestar de diferentes maneiras, afetando o fígado, o sistema nervoso e até o comportamento das pessoas.
O gene ATP7B é responsável por ajudar o fígado a remover o excesso de cobre do corpo. Quando esse gene apresenta defeito, o fígado não consegue realizar essa tarefa corretamente. Como resultado, o cobre começa a se acumular dentro das células do fígado, chamadas hepatócitos (LI; WANG; CHEN, 2023).
Inicialmente, esse acúmulo de cobre não provoca sintomas, mas com o tempo, o cobre vai se acumulando cada vez mais até atingir níveis que começam a prejudicar as células do fígado. Quando a capacidade de armazenamento hepático é excedida, o cobre é liberado para a circulação, se espalhando em outros órgãos como cérebro, rins e córnea. Esse acúmulo extracelular é responsável pelas manifestações neurológicas e psiquiátricas observadas nos pacientes (PFEIFFER, 2007). De acordo com Ala et al. (2007), “o acúmulo cerebral de cobre afeta principalmente os gânglios da base, estruturas cruciais para o controle motor, resultando em tremores, distonia e parkinsonismo”.
Manifestações Clínicas
A apresentação clínica é heterogênea, variando desde quadros assintomáticos até formas graves. Em crianças predominam as manifestações hepáticas, enquanto em adolescentes e adultos jovens são mais comuns os sintomas neurológicos e psiquiátricos (PFEIFFER, 2007).
Manifestações hepáticas
As manifestações hepáticas são geralmente as primeiras alterações a serem observadas na Doença de Wilson, especialmente em crianças e jovens. Elas podem se apresentar de forma assintomática, com elevações discretas de enzimas hepáticas, ou evoluir para quadros mais graves como hepatite crônica, cirrose e insuficiência hepática fulminante.
A hepatite fulminante relacionada à Doença de Wilson é particularmente grave e pode ocorrer em adolescentes e jovens adultos, sendo frequentemente associada a anemia hemolítica não imune. De acordo com Socha et al. (2018), “apresentação hepática pode variar desde anormalidades laboratoriais discretas até falência hepática aguda, sendo necessário a suspeita clínica precoce para a instituição de medidas terapêuticas adequadas”.
Pfeiffer (2007) complementa que:
“A insuficiência hepática fulminante em pacientes jovens, especialmente na ausência de causa viral identificável, deve sempre levantar a suspeita de Doença de Wilson. A falta de reconhecimento dessa associação pode levar ao atraso no transplante hepático, que muitas vezes é a única medida de salvamento nesses casos” (PFEIFFER, 2007, p. 125).
Manifestações neurológicas
As manifestações neurológicas da Doença de Wilson surgem tipicamente após o início da disfunção hepática, refletindo o acúmulo de cobre no sistema nervoso central, especialmente nos gânglios da base. Os sintomas neurológicos são variados e incluem tremores, rigidez, disartria, distonia e bradicinesia. Essas manifestações podem ser incapacitantes e muitas vezes, são confundidas com outras doenças neurológicas degenerativas, como a doença de Parkinson.
Conforme Członkowska et al. (2018), “os distúrbios motores, especialmente a rigidez e o tremor, refletem o acúmulo de cobre nos gânglios da base”. Ferenci et al. (2019) destacam que:
“Os sintomas neurológicos da Doença de Wilson podem se manifestar de forma insidiosa, inicialmente como dificuldades de coordenação ou alterações sutis da fala, progredindo para tremores incapacitantes, distonia severa e rigidez generalizada. O reconhecimento precoce desses sinais é fundamental, pois a progressão do dano neurológico pode ser parcialmente revertida com o tratamento adequado, embora a recuperação completa seja rara em casos avançados” (FERENCI et al.,2019, p. 165).
Manifestações psiquiátricas
Alterações psiquiátricas podem preceder ou acompanhar as manifestações neurológicas e hepáticas. Os sintomas incluem mudanças de comportamento, irritabilidade, depressão, ansiedade, declínio cognitivo e, em casos mais graves, episódios psicóticos. Essas manifestações são frequentemente subestimadas ou atribuídas a transtornos psiquiátricos primários, atrasando o diagnóstico correto (ROBERTS; SCHILSKY, 2008). Pfeiffer (2007) ressalta que “o reconhecimento das alterações comportamentais é crucial para o diagnóstico precoce”.
Alterações oftalmológicas
O anel de Kayser-Fleischer, visualizado através do exame com lâmpada de fenda, é um achado oftalmológico característico da Doença de Wilson. Ele representa a deposição de cobre na membrana de Descemet da córnea e é altamente prevalente em pacientes com manifestações neurológicas (ALA et al., 2007). Segundo Pfeiffer (2007), “a presença do anel de Kayser-Fleischer é observada em quase todos os pacientes com manifestações neurológicas da doença”.
Manifestações hematológicas
Distúrbios hematológicos, como anemia hemolítica Coombs negativa, são manifestações iniciais importantes da Doença de Wilson. Esse fenômeno ocorre devido ao efeito citotóxico do cobre livre circulante nas membranas dos eritrócitos, levando à lise celular (FERENCI et al., 2019).
Alterações renais e ósseas
A Doença de Wilson também pode afetar os rins, manifestando-se como síndrome de Fanconi, caracterizada por perda de glicose, fosfato, bicarbonato e aminoácidos na urina. O comprometimento ósseo, como osteopenia e artrite, também pode ocorrer devido à deposição anormal de cobre nos ossos e articulações (CZŁONKOWSKA; LITWIN; DUSEK, 2018). De acordo com Członkowska et al (2018):
“As manifestações renais da Doença de Wilson, embora menos frequentes, podem ser clinicamente significativas, incluindo proteinúria, síndrome de Fanconi e, em casos avançados, insuficiência renal progressiva. A osteopenia, frequentemente observada em pacientes não tratados, resulta tanto da toxicidade direta do cobre sobre os osteoblastos quanto da disfunção renal associada” (CZŁONKOWSKA; LITWIN; DUSEK, 2018, p. 1430).
Manifestações cardíacas
Embora raras, podem ocorrer complicações cardíacas, como cardiomiopatia dilatada, arritmias e disfunções autonômicas. Essas manifestações resultam da deposição de cobre no miocárdio e estão associadas a pior prognóstico em pacientes com Doença de Wilson avançada (ROBERTS; SCHILSKY, 2008).
Abaixo, segue quadro comparativo:
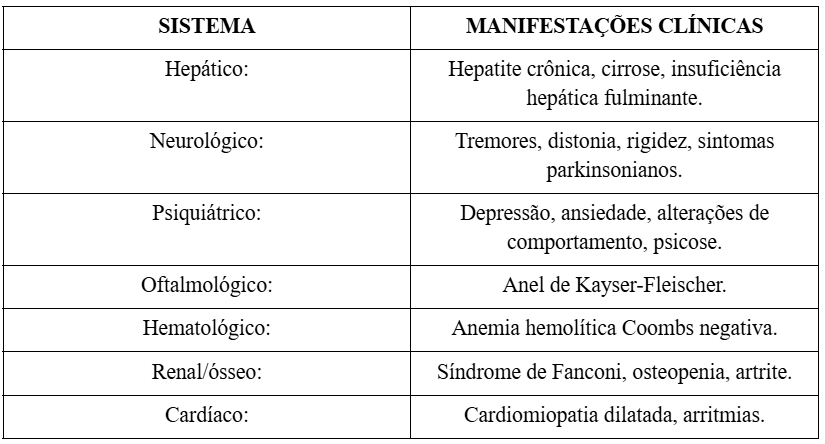
Diagnóstico
O diagnóstico é baseado em critérios clínicos, laboratoriais e genéticos, pois não há exame único definitivo. Inclui: ceruloplasmina sérica reduzida (20 mg/dl), cupremia de 24h (100 µg/24h em adultos), cobre hepático elevado na biópsia (250 µg/g de peso seco), exame oftalmológico para detecção do anel de Kayser-Fleischer, e testes genéticos para mutação do ATP7B. Os critérios de Leipzig combinam esses achados, aumentando a acurácia diagnóstica (ROBERTS; SCHILSKY, 2008).
A relação entre a Doença de Wilson e a baixa concentração de ceruloplasmina no sangue foi descoberta em 1952, através de estudos feitos ao mesmo tempo por Scheinberg e Gitlin, e por Bearn e Kunkel (SCHEINBERG; GITLIN, 1952; BEARN; KUNKEL, 1952). Essa descoberta foi muito importante, pois tornou possível o desenvolvimento de exames laboratoriais específicos para ajudar no diagnóstico da doença. Em indivíduos saudáveis, a maior parte do cobre circulante está ligada à ceruloplasmina. Na Doença de Wilson, a deficiência dessa proteína leva ao aumento do cobre livre no plasma, que é altamente tóxico para os tecidos embora sua ausência isolada não seja suficiente para o diagnóstico (SCHEINBERG; GITLIN, 1952, p. 875).
Logo, ceruloplasmina sérica quando apresenta valores normais em alguns casos, torna esse exame um marcador de utilidade limitada quando isolado. Assim, seu resultado deve ser interpretado em conjunto com outros testes laboratoriais e com a avaliação clínica do paciente. A dependência exclusiva da dosagem de ceruloplasmina pode atrasar o diagnóstico, especialmente em pacientes com manifestações atípicas ou em estágios iniciais da doença, reforçando a necessidade de uma abordagem diagnóstica mais abrangente.
Com o avanço da biologia molecular, foi identificado o gene ATP7B como o principal responsável pela Doença de Wilson (FERENCI et al., 2019). Esse gene é importante porque comanda a produção de uma proteína que regula o transporte de cobre no corpo. Quando há alguma alteração nesse gene, o organismo não consegue eliminar o cobre de forma adequada, levando ao seu acúmulo nos órgãos, principalmente no fígado e no cérebro.
A presença do anel de Kayser-Fleischer é observada em cerca de 95% dos pacientes com manifestações neurológicas. O exame com lâmpada de fenda é um método não invasivo e altamente útil. Pfeiffer (2007) afirma que “a detecção do anel é um forte indicativo de Doença de Wilson, principalmente em pacientes neurológicos”.
Tratamento
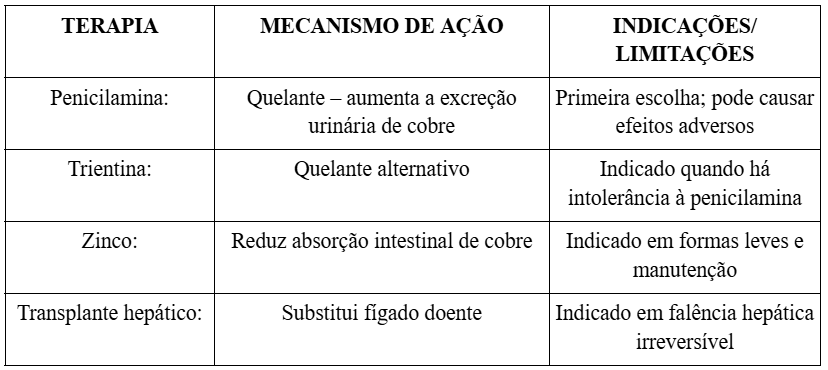
Prognóstico
O prognóstico está diretamente relacionado à precocidade do diagnóstico e adesão ao tratamento. Pacientes diagnosticados precocemente podem alcançar qualidade de vida normal. Já diagnósticos tardios frequentemente evoluem com sequelas neurológicas irreversíveis ou necessidade de transplante hepático (FERENCI et al., 2019).
3. METODOLOGIA
Este estudo caracteriza-se como uma revisão sistemática da literatura. O protocolo metodológico seguiu as etapas de busca, seleção, extração e análise dos dados dos estudos incluídos, com o objetivo de compreender os impactos clínicos e terapêuticos do diagnóstico tardio da Doença de Wilson (DW). A escolha dessa abordagem metodológica justifica-se pela necessidade de aprofundar o conhecimento sobre uma condição rara e complexa, cuja detecção precoce é fundamental para a eficácia do tratamento.
A amostra do estudo foi composta principalmente por artigos publicados entre 2007 e 2024, priorizando-se revisões sistemáticas e estudos clínicos recentes disponíveis nas bases PubMed, SciELO e Google Acadêmico. Contudo, também foram incluídas fontes relevantes anteriores a esse período, quando estas continham fundamentações teóricas essenciais ou histórico clínico consolidado sobre a Doença de Wilson. Os critérios de inclusão abrangeram publicações em inglês e português que abordassem aspectos clínicos, diagnósticos e terapêuticos da DW, especialmente relacionados ao diagnóstico tardio.
Para definir a amostragem de artigos científicos, foi realizada uma análise criteriosa das publicações mais relevantes sobre o tema, com base em sua qualidade metodológica, atualidade e relevância para os objetivos do estudo. A seleção de fontes foi feita de acordo com a contribuição teórica ou prática que os estudos poderiam oferecer à compreensão dos efeitos do diagnóstico tardio e das abordagens terapêuticas.
Tipo de publicação: Artigos científicos de revisão de estudos clínicos, e trabalhos que abordam a Doença de Wilson, com foco no diagnóstico tardio, manifestações clínicas e opções terapêuticas.
Idioma: Apenas artigos em português e inglês foram considerados.
Qualidade metodológica: Foram selecionados estudos com rigor metodológico comprovado, com descrição clara dos métodos, amostragem e resultados. Artigos que se mostraram inconsistentes ou com falhas em sua abordagem metodológica foram excluídos.
4. RESULTADOS E DISCUSSÕES
Os resultados obtidos a partir da revisão bibliográfica evidenciam que o diagnóstico tardio da Doença de Wilson (DW) está fortemente associado à diversidade de suas manifestações clínicas e à dificuldade de interpretação dos exames laboratoriais iniciais. Essa heterogeneidade sintomática — que pode envolver alterações hepáticas, neurológicas, psiquiátricas e oftalmológicas — leva frequentemente à confusão diagnóstica com outras doenças hepáticas ou distúrbios neurológicos.
De acordo com Ferenci et al. (2019), a faixa etária entre 5 e 35 anos representa o período de maior incidência da DW, e a falta de suspeita clínica em pacientes jovens com sintomas hepáticos atípicos é uma das principais causas de atraso no diagnóstico. Socha et al. (2018) reforçam que, em crianças e adolescentes, o quadro inicial pode se limitar a discreta elevação das enzimas hepáticas, sendo facilmente confundido com hepatites virais ou doenças metabólicas benignas. Já em adultos jovens, as manifestações neurológicas e psiquiátricas costumam predominar, levando a diagnósticos equivocados de distúrbios de movimento e até mesmo Parkinson precoce ou transtornos mentais primários (PFEIFFER, 2007; SILVA; PEREIRA, 2022).
Os estudos de Scheinberg e Gitlin (1952) e Linder (2018) mostram que, embora a dosagem de ceruloplasmina seja um marcador importante, sua interpretação isolada pode ser enganosa, visto que valores normais não excluem a DW. Assim, a combinação entre ceruloplasmina reduzida, cupremia de 24 horas elevada e presença do anel de Kayser- Fleischer é considerada a base diagnóstica mais confiável (ALA et al., 2007; ROBERTS; SCHILSKY, 2008). Ainda assim, o diagnóstico definitivo muitas vezes depende da biópsia hepática, que quantifica o conteúdo de cobre no fígado (CZŁONKOWSKA; LITWIN; DUSEK, 2018).
Em relação ao tratamento, os resultados analisados destacam a importância do início precoce da terapia quelante para prevenir complicações irreversíveis. O uso de penicilamina, trientina e zinco se mostrou eficaz em controlar a sobrecarga de cobre quando instituído antes do surgimento de cirrose ou alterações neurológicas graves (PFEIFFER, 2007; FERENCI et al., 2019; SAMPAIO et al., 2023). Entretanto, nos casos em que o diagnóstico é tardio, o dano hepático é frequentemente irreversível, tornando o transplante hepático a única chance de uma sobrevida.
Os achados também apontam que a ausência de protocolos padronizados de triagem para doenças raras, aliada à escassa capacitação de profissionais da atenção primária, agrava o subdiagnóstico da DW (FERREIRA; OLIVEIRA, 2021). A European Association for the Study of the Liver (EASL, 2012) recomenda a inclusão da DW no diagnóstico diferencial de qualquer hepatopatia inexplicada em jovens, o que ainda não é rotina em muitos centros brasileiros. Tal lacuna reforça a necessidade de políticas públicas de conscientização.
Além dos impactos clínicos, o atraso no diagnóstico gera consequências psicossociais importantes. Os pacientes frequentemente passam por longos períodos de sofrimento devido à incerteza diagnóstica e à deterioração progressiva da saúde física e mental (NASCIMENTO; OLIVEIRA, 2020). Essa situação reforça a importância de uma abordagem multidisciplinar, envolvendo hepatologistas, neurologistas, geneticistas e psicólogos, para garantir um manejo integral da doença.
Portanto, a análise dos estudos confirma que o diagnóstico precoce é um fator determinante do prognóstico favorável na Doença de Wilson. A implementação de protocolos clínicos que integrem testes bioquímicos e genéticos, aliados à capacitação dos profissionais de saúde, pode reduzir significativamente a taxa de diagnósticos tardios, melhorando o tratamento e a qualidade de vida dos pacientes acometidos.
5. CONCLUSÃO/CONSIDERAÇÕES FINAIS
O estudo confirma que a identificação precoce da doença é um fator de sucesso terapêutico. A instituição imediata de tratamento quelante ou terapia com sais de zinco é capaz de evitar a progressão dos sintomas e proporcionar uma qualidade de vida praticamente normal ao paciente (PFEIFFER, 2007; SAMPAIO et al., 2023). No entanto, quando o diagnóstico ocorre em estágios avançados, o transplante hepático se torna, muitas vezes, a única opção de sobrevida, reforçando a necessidade de políticas de triagem mais eficazes (SOCHA et al., 2018).
Além dos impactos clínicos, observou-se que o atraso no diagnóstico gera também consequências sociais e psicológicas importantes, devido à longa jornada até a confirmação da doença e à limitação funcional progressiva dos pacientes (NASCIMENTO; OLIVEIRA, 2020). Assim, torna-se imprescindível que os profissionais de saúde, principalmente os da atenção básica, estejam capacitados para reconhecer precocemente os sinais e sintomas sugestivos da DW.
Conclui-se que o diagnóstico precoce da Doença de Wilson é um fator essencial para o sucesso terapêutico e a melhoria da qualidade de vida dos pacientes. Por tratar-se de uma revisão sistemática, este estudo fornece uma visão abrangente e baseada em evidências sobre os impactos do diagnóstico tardio e as estratégias terapêuticas da DW. A revisão destaca a importância da capacitação profissional e da padronização dos protocolos diagnósticos como medidas prioritárias para reduzir a morbimortalidade associada à doença.
Por fim, este trabalho contribui para o campo da Biomedicina ao reforçar a importância do diagnóstico precoce como ferramenta determinante de prognóstico e ao destacar a necessidade de integração entre pesquisa científica, prática clínica e educação continuada como estratégias para melhorar a detecção e o manejo da Doença de Wilson.
6. REFERÊNCIAS
ALA, A. et al. Wilson’s disease. The Lancet, v. 369, n. 9559, p. 397–408, 2007.
BEARN, A. G.; KUNKEL, H. G. Abnormalities of copper metabolism in Wilson’s disease and in other chronic hepatic diseases. Journal of Clinical Investigation, v. 31, n. 5, p. 558–568, 1952.
CARTAXO, T. A. Doença de Wilson: aspectos clínicos, diagnósticos e terapêuticos. Revista Brasileira de Neurologia, v. 57, n. 3, p. 45–52, 2021.
CZŁONKOWSKA, A. et al. Wilson’s disease. Nature Reviews Disease Primers, v. 4, n. 21, p. 1–20, 2018.
CZŁONKOWSKA, A.; LITWIN, T.; DUSEK, P. Clinical spectrum and management of Wilson disease in different populations. Journal of Hepatology, v. 68, n. 6, p. 1430–1441, 2018.
DOOLEY, J. S. Wilson disease: historical and clinical perspectives. Hepatology International, v. 18, n. 1, p. 15–24, 2024.
EASL – EUROPEAN ASSOCIATION FOR THE STUDY OF THE LIVER. EASL Clinical Practice Guidelines: Wilson’s disease. Journal of Hepatology, v. 56, n. 3, p. 671–685, 2012.
FERENCI, P. et al. Diagnosis and phenotypic classification of Wilson disease. Liver International, v. 39, n. 1, p. 162–170, 2019.
FERREIRA, L. M.; OLIVEIRA, C. R. Subdiagnóstico de doenças raras no Brasil: desafios e perspectivas. Revista de Saúde Coletiva, v. 31, n. 2, p. 112–119, 2021.
LI, Y.; WANG, J.; CHEN, X. Molecular mechanisms and genetic basis of Wilson’s disease. Frontiers in Genetics, v. 14, p. 101–110, 2023.
LINDER, M. C. Copper metabolism and its disorders: recent insights. Nutrients, v. 10, n. 8, p. 1–14, 2018.
NASCIMENTO, F. M.; OLIVEIRA, P. J. Aspectos psicossociais do diagnóstico tardio em doenças genéticas raras. Revista Psicologia e Saúde, v. 12, n. 1, p. 25–38, 2020.
PFEIFFER, R. F. Wilson’s Disease. 3. ed. Philadelphia: Elsevier, 2007.
ROBERTS, E. A.; SCHILSKY, M. L. Diagnosis and treatment of Wilson disease: an update. Hepatology, v. 47, n. 6, p. 2089–2111, 2008.
SAMPAIO, A. C. et al. Tratamento farmacológico da Doença de Wilson: avanços e desafios. Revista Brasileira de Medicina, v. 80, n. 4, p. 221–230, 2023.
SCHEINBERG, I. H.; GITLIN, D. Deficiency of ceruloplasmin in patients with hepatolenticular degeneration (Wilson’s disease). Science, v. 116, n. 3018, p. 484–485, 1952.
SILVA, L. F.; PEREIRA, R. M. Manifestações neuropsiquiátricas da Doença de Wilson: revisão narrativa. Arquivos de Neuro-Psiquiatria, v. 80, n. 2, p. 130–138, 2022.
SOCHA, P. et al. Wilson’s disease in children: a review of clinical presentation and therapy. Journal of Pediatric Gastroenterology and Nutrition, v. 66, n. 3, p. 345–356, 2018.
1Univel – Centro Universitário de Cascavel