IMMUNOMETABOLIC DYSFUNCTION IN ALZHEIMER’S DISEASE: AUTOIMMUNE PARADIGM
DISFUNCIÓN INMUNOMETABÓLICA EN LA ENFERMEDAD DE ALZHEIMER: PARADIGMA AUTOINMUNE
REGISTRO DOI: 10.69849/revistaft/th102502201540
Rosa Maria Braga Lopes de Moura*
Cristiane Rodigues da Silva
Daiana Maich Mathias
Francine Trindade Soares
Gislene Gomes das Neves
Marina Costa
Mariana Machado Fernandes
Nicolau Pereira
Rejane Magano Souza
Tainara Kauffmann Machado
RESUMO
Evidências emergentes indicam que a função metabólica desempenha um papel crítico na regulação da função microglial na Doença de Alzheimer (DA) com programação metabólica subjacente a diversas funções e fenótipos imunológicos microgliais. Recentemente, as evidências sugerem que a DA é um distúrbio imunometabólico generalizado no qual o metabolismo celular alterado ocorre nos estágios iniciais da doença. Desse modo, a mudança na dependência metabólica não resultou apenas em capacidades glicolíticas cerebrais prejudicadas, mas também em aminoácidos que são essenciais para a reciclagem e síntese de neurotransmissores com comprometimento na fidelidade sináptica, que é a base molecular das funções cognitivas e de memória. Diante do exposto, o objetivo do presente estudo foi avaliar o novo modelo de DA intitulada como doença autoimune. Para tanto, foi realizada uma revisão integrativa da literatura com os descritores “Alzheimer como Doença Autoimune”, “Endofenótipo imunometabólico”, “Disfunção mitocondrial na DA” bem como “Disfunção Imunometabólica na DA” nas bases de dados PubMed. Os resultados apontam que o sistema imunológico inato é ativado, levando à liberação de Aβ como um imunopeptídeo que subsequentemente inflige um ataque mal direcionado aos neurônios do hospedeiro – um evento autoimune.
Palavras-chave: DA, Endofenótipo, Imunometabolismo, Autoimune, Mitofagia.
ABSTRACT
Emerging evidence indicates that metabolic function plays a critical role in regulating microglial function in Alzheimer’s Disease (AD) with metabolic programming underlying diverse microglial immune functions and phenotypes. Recently, evidence suggests that AD is a widespread immunometabolic disorder in which altered cellular metabolism occurs in the early stages of the disease. Thus, the change in metabolic dependence not only resulted in impaired brain glycolytic capabilities, but also in amino acids that are essential for the recycling and synthesis of neurotransmitters with compromised synaptic fidelity, which is the molecular basis of cognitive and memory functions. In view of the above, the objective of the present study was to evaluate the new model of AD called an autoimmune disease. To this end, an integrative review of the literature was carried out using the descriptors “Alzheimer as an Autoimmune Disease”, “Immunometabolic Endophenotype”, “Mitochondrial Dysfunction in AD” as well as “Immunometabolic Dysfunction in AD” in the PubMed databases. The results indicate that the innate immune system is activated, leading to the release of Aβ as an immunopeptide that subsequently inflicts a misdirected attack on the host’s neurons – an autoimmune event.
Keywords: AD, Endophenotype, Immunometabolism, Autoimmune, Mitophagy.
RESUMEN
La evidencia emergente indica que la función metabólica desempeña um papel fundamental em la regulación de la función microglial em la enfermedad de Alzheimer (EA), com uma programación metabólica subyacente a diversas funciones y fenotipos inmunológicos microgliales. Recientemente, la evidencia sugiere que la EA es um trastorno inmunometabólico generalizado em el que se produce uma alteración del metabolismo celular em las primeras etapas de la enfermedad. Así, el cambio em la dependencia metabólica no sólo resultó em uma alteración de las capacidades glucolíticas del cerebro, sino también em aminoácidos esenciales para el reciclaje y la síntesis de neurotransmisores com uma fidelidad sináptica comprometida, que es la base molecular de las funciones cognitivas y de la memoria. Em vista de lo anterior, el objetivo del presente estudio fue evaluar el nuevo modelo de EA denominado enfermedad autoinmune. Para ello, se realizó uma revisión integradora de la literatura utilizando los descriptores “Alzheimer as na Autoimmune Disease”, “Immunometabolic Endophenotype”, “Mitochondrial Dysfunction in AD” así como “Immunometabolic Dysfunction in AD” em las bases de datos PubMed. Los resultados indican que el sistema inmunológico innato se activa, lo que lleva a la liberación de Aβ como um inmunopéptido que posteriormente inflige um ataque mal dirigido a las neuronas del huésped, um evento autoinmune. Palabras clave: EA, Endofenotipo, Inmunometabolismo, Autoinmune, Mitofagia
INTRODUÇÃO
A doença de Alzheimer (DA) foi descrita pelo médico alemão Alois Alzheimer, em 1906 caracterizando como uma patologia neurológica, de etiologia desconhecida e de aparecimento insidioso. Segundo o relatório da Organização Mundial da Saúde (OMS), o número de pessoas com demência cresce em todo o mundo, estimando-se 78 milhões em 2030 e 139 milhões em 2050.
O Ministério da Saúde divulgou em 2024, o Relatório Nacional sobre a Demência: Epidemiologia, (re)conhecimento e projeções futuras, que conta com dados epidemiológicos, risco, subdiagnóstico e estigma dessa condição. De acordo com o estudo, cerca de 8,5% da população com 60 anos ou mais convivem com a doença, representando um número aproximado de 2,71 milhões de casos. Até 2050, a projeção é que 5,6 milhões de pessoas sejam diagnosticadas no país. As informações foram anunciadas durante evento realizado na sede da Organização Pan-Americana da Saúde (Opas), em Brasília.
A prevalência aumenta com a idade, sendo que cerca de 1% das pessoas com 65 anos ou mais têm DA. Estima-se que sua incidência duplica a cada 5 anos após essa idade. um distúrbio neurodegenerativo altamente prevalente, responsável por 60–80% dos casos de demência e classificado como a sexta maior causa de morte no mundo. O número total estimado de pessoas que sofrem de demência em todo o mundo aumentou para perto de 50 milhões, com um custo de mais de 1 bilião de dólares anuais (GUSTAVSSON et al., 2023).
A fase pré-clínica da DA pode começar 20 anos antes do diagnóstico, o que proporciona uma janela alargada para medidas preventivas que exerceriam um impacto transformador na incidência e prevalência da DA. A crescente prevalência da DA exige uma exploração abrangente da sua etiologia complexa, com foco na vulnerabilidade específica do sexo, particularmente na maior suscetibilidade observada em mulheres na pós-menopausa. Alterações neurometabólicas durante a transição endócrina emergem como indicadores precoces da patologia da DA, incluindo redução do metabolismo da glicose e aumento da deposição de beta-amilóide (Aβ). O ambiente endócrino flutuante, marcado pelo declínio dos níveis de estradiol e pela redução da atividade do receptor beta de estrogênio (ERβ), agrava ainda mais esse processo. Neste contexto, o Forkhead O3 (FOXO3) é um mediador crítico que liga distúrbios metabólicos ao declínio hormonal desempenhando um papel fundamental na intersecção da menopausa e da DA, dada a sua desregulação tanto em pacientes com DA como em mulheres na pós-menopausa. Esta relação destaca a interseção entre alterações hormonais e aumento da suscetibilidade à DA abrindo uma discussão sobre a contribuição do FOXO3 para a desregulação metabólica observada na menopausa e seu impacto na progressão da DA (O’MAHONY, 2025).
Estudos epidemiológicos de homens de meia-idade e acima de 60 anos demonstraram associações de concentrações mais baixas de testosterona com maior prevalência e incidência de declínio cognitivo e demência, incluindo a DA. Em estudos observacionais, homens com cancro da próstata tratados com terapia de privação androgénica apresentaram um risco mais elevado de demência. Pequenos estudos de intervenção sobre testosterona utilizando diferentes medidas da função cognitiva forneceram resultados inconsistentes (YEAP, 2025).
Os fatores de risco incluem menor escolaridade, tabagismo, obesidade, consumo excessivo de álcool, traumatismo cranioencefálico, hipertensão, deficiência auditiva, depressão, poluição do ar, inatividade física, diabetes e baixo contato social (BIANCHI, 2025; LIVINGSTON, 2025).
De acordo com o CID11 6D80 DSM-V, a DA começa por meio do diagnóstico de um transtorno neurocognitivo maior ou leve, que inclui o quadro de síndrome demencial com início gradual e declínio cognitivo contínuo. Ele exclui o uso de substâncias ou outras condições que mimetizam a doença.
A perda episódica de memória de curto prazo é o sintoma inicial e mais comum da DA típica. Os indivíduos podem ter dificuldade em reter novas informações enquanto ainda recordam memórias de longo prazo apresentando deficiências na resolução de problemas, no julgamento, no funcionamento executivo, nas habilidades organizacionais e pensamento abstrato. Nos estágios iniciais da doença, os comprometimentos do funcionamento executivo podem variar de sutis a significativos. As atividades instrumentais da vida diária como dirigir, administrar finanças, cozinhar e planejar atividades detalhadas, são afetadas relativamente cedo na demência. Esses primeiros sinais de declínio cognitivo são seguidos por distúrbios de linguagem e comprometimento das habilidades visuoespaciais. Sintomas neuropsiquiátricos como apatia, retraimento social, desinibição, agitação, psicose e perambulação também são comuns nos estágios moderados a tardios (KUMAR, 2024).
A doença é classificada como familiar e esporádica. A apresentação familiar dominante ou autossômica representa 1-5% do número total de casos. É categorizada como de início precoce (EOAD; <65 anos de idade) e apresenta mutações genéticas na presenilina 1 (PSEN1), presenilina 2 (PSEN2) ou na proteína precursora amilóide (APP). A DA esporádica representa 95% dos casos e é categorizada como de início tardio (LOAD), ocorrendo em pacientes com mais de 65 anos de idade. O fator de risco identificado na DA esporádica: o envelhecimento (ANDRADE-GUERRERO, 2024).
Estudos genômicos apontam uma mutação no gene TREM (myeloidcells) com a substituição na proteína de uma histidina por uma arginina, que confere risco para DA tanto quanto o APOE-e4. Estudos posteriores apontam que os genes PSEN1 e PSEN 2, alteram o padrão de clivagem da beta secretase ocasionando a sua produção anormal. Além disso, o alelo E4 do gene da APOE foi identificado como o principal fator de risco genético no desenvolvimento da forma esporádica da DA (HOOGMARTENS, 2021).
Os tratamentos farmacológicos são os anticolinesterásicos (inibidores da enzima acetilcolinesterase (AChE) e um antagonista do receptor N-metil-D-aspartato (NMDA) (CUMMINGS, 2019). Os anticolinesterásicos como donepezila, rivastigmina e galantamina inibem a enzima AChE aumentando a disponibilidade de acetilcolina na fenda sináptica (HAAKE et al., 2020).
A memantina é um antagonista não-competitivo de receptores NMDA, permitindo sua ativação fisiológica durante os processos de formação da memória, porém bloqueando a abertura dos canais e sua ativação patológica. Por ser um antagonista do receptor glutamatérgico NMDA, o fármaco atua bloqueando os receptores NMDA e inibindo o influxo excessivo de Ca2+, assim, evitando que ocorra a excitotoxicidade e consequente morte dos neurônios de glutamato (WANG, 2017).
A disfunção glutamatérgica consiste no fato de que o glutamato é o principal neurotransmissor excitatório do Sistema Nervoso Central (SNC). Este neurotransmissor é mediado por receptores ionotrópicos [N-metil-D-aspartato (NMDA), [ácido α-amino-3-hidroxi-5-metil-4- isoxazolepropiônico (AMPA) e o cainato (CAR) e metabotrópicos. Na DA, ocorre a ativação excessiva de receptores ionotrópicos, especialmente o NMDA, levando a alteração da homeostase de cálcio e, consequentemente, aumentando sua concentração intracelular, originando um processo de apoptose e neurodegeneração (OERTEL, 2016).
O complexo colinérgico basal do cérebro, constituído pelo septo medial, banda diagonal horizontal e vertical de Broca e o núcleo basal de Meynert, fornece as projeções colinérgicas do córtex cerebral e hipocampo. Presume-se que os neurônios colinérgicos deste complexo sofram alterações neurodegenerativas moderadas durante o envelhecimento, no entanto, a perda neuronal de células foi predominante em processos patológicos, como na DA, sendo amplamente documentada uma grave perda de inervação colinérgica cortical. Portanto, a função colinérgica é importante no processo de memória e de aprendizagem. Desse modo, a hipótese colinérgica relaciona a DA com a diminuição da concentração da colina acetiltransferase (AChT), que é a enzima responsável pela produção de acetilcolina (ACh), no hipocampo e no córtex (FALCO, 2015).
A micróglia desempenha múltiplas funções no sistema nervoso central, incluindo vigilância, fagocitose e liberação de uma variedade de fatores solúveis. É importante ressaltar que a maioria de suas funções está intimamente relacionada a mudanças em seu metabolismo. Essa interdependência natural entre as principais propriedades microgliais e o metabolismo oferece uma oportunidade única de modular as atividades microgliais por meio de intervenções nutricionais ou metabólicas Em contrapartida, a fagocitose descontrolada e não seletiva de sinapses saudáveis pela microglia pode contribuir para a neurodegeneração (VILALTA, 2018 apud LEPIARZ-RABA, 2023).
A homeostase fisiológica é um equilíbrio entre a biogênese das mitocôndrias funcionais e a degradação autofágica das disfuncionais através de um processo seletivo denominado mitofagia. Durante esta via de limpeza, as mitocôndrias prejudiciais são engolfadas por uma membrana dupla (fagóforo) formando o mitofagossoma, que então se fundirá com um lisossoma para formar um mitofagolisossomo. Nesta estrutura, as hidrolases lisossomais irão digerir asmitocôndrias em pequenos componentes que podem então ser reciclados. Estudos identificaram dois tipos principais de mitofagia (PINK1/Parkin-dependente ou independente) ativada por uma infinidade de estímulos e implicando diferentes proteínas exibindo receptores de mitofagia (MARY, 2023).
A resposta aberrante da micróglia aos depósitos de Aβ e neurônios em degeneração pode levar a estados inflamatórios que contribuem ainda mais para danos neuronais. Assim, encontrar maneiras de aumentar preferencialmente a fagocitose microglial de depósitos tóxicos como Aβ, sem degradação de sinapses saudáveis ou respostas inflamatórias exageradas, pode ser uma estratégia preventiva e terapêutica eficaz (LEPIARZ-RABA, 2023).
Os indivíduos com a DA apresentam níveis plasmáticos reduzidos de triptofano e a depleção aguda de triptofano causa aumento da disfunção cognitiva. O importante papel do microbioma intestinal na produção de metabólitos neuroativos representa uma nova abordagem para intervenção terapêutica (MEIER‐STEPHENSON, 2022 apud MOURA, 2024). Dessas observações emergem múltiplas abordagens terapêuticas.
O vertiginoso desenvolvimento de pesquisas em engenharia genética e em medicina biomolecular – fundamentalmente com o advento de novas técnicas de DNA / RNA recombinantes – com vistas, dentre outros, a identificar e tratar as doenças de ensejo a um “trio” de possíveis ações: fala-se em (i) conhecer, (ii) predizer e (iii) mudar.
Os estudos experimentais oscilaram entre conquistas e fracassos. Não obstante, o número de protocolos e de ensaios pré-clínicos e clínicos aprovados tem aumentado significativamente, o que salienta a necessidade de se pôr em pauta todas as alternativas possíveis. A fase de desenvolvimento desses tratamentos permite ainda uma última dúvida: saber se já se está no campo efetivo de uma terapia inovadora ou se ainda se caminha pelo palco da investigação e da experimentação. Dada a grande maioria dos projetos ainda se encontrar em uma prematura fase de testes (ARCHER, 1997).
O modelo intitulado “Alzheimer como Doença Autoimune” (AD2) postula a DA como uma doença autoimune de imunidade inata e não de imunidade adaptativa. Convencionalmente, qualquer lesão não intencional associada à disfunção imune inata é considerada autoinflamatória em vez de autoimune, surgindo principalmente como um efeito não específico de um estado pró-inflamatório. No entanto, como conjecturado em AD2, a DA é uma doença autoimune de imunidade inata porque Aβ liberada como antibacteriano “confunde” os neurônios ao interpretar como não-próprio, perfurando a sua membrana como faria com uma bactéria (WEIVER, 2023).
O modelo supracitados, representa uma conceituação inovadora da DA como uma doença autoimune e autoinflamatória centrada no cérebro da imunidade inata. O modelo AD2 se esforça para harmonizar outras proposições mecanicistas (incluindo proteopatia, sinaptotoxicidade e mitocondriopatia), ao mesmo tempo que reconhece Aβ como um imunopeptídeo fisiologicamente oligomerizante e parte de um processo imunopático muito maior, abrangente e altamente interligado.
Contrariamente à hipótese da cascata amilóide, a proteína Aβ pode ser protetora para os neurônios do SNC nas fases iniciais de eventos de estresse. Por outro viés, quando os estressores crônicos persistem é que a proteína Aβ se torna superexpressa e forma oligômeros e protofibrilas com toxicidade. Assim, as alterações fisiopatológicas que ocorrem no ambiente do SNC e que induzem estresse neuronal crônico explica a heterogeneidade na trajetória da doença dependendo da combinação de comorbidades subjacentes em um paradigma de doença autoimune.
AUTOFAGIA
A autofagia é a principal via de degradação lisossômica, mantendo a homeostase celular ao transformar constitutivamente proteínas e organelas obsoletas. É ainda induzido por doenças e estresse celular para eliminar proteínas anormais, agregados e organelas danificadas (BOLAND, 2018; MENZIES, 2017; MIZUSHIMA, 2021).
O Aβ é importado para as mitocôndrias. Uma vez dentro da matriz mitocondrial, a protease de pré-sequência de metaloprotease (PreP) de 110 kDa é capaz de degradar Aβ para reduzir os efeitos tóxicos no funcionamento mitocondrial (WAN, 2017; PINTO, 2014). Dadas as diversas formas de degradação do Aβ, pode-se inferir que a disfunção mitocondrial pode resultar no aumento dos níveis de Aβ em todo o corpo, o que poderia levar ao desenvolvimento de distúrbios crônicos e aumentar o estresse neuronal.
Além da deposição de Aβ e da agregação de tau, a disfunção sináptica é outra característica da DA. Sinapses são estruturas específicas de neurônios que servem como unidades básicas para a comunicação de neurônios pré-sinápticos para pós-sinápticos. Foi observado que o número de sinapses é reduzido na fase inicial da patogênese da DA (Chen et al., 2019). Recentemente, evidências propõem que a autofagia funcional é necessária para funções sinápticas, incluindo neurotransmissão e plasticidade sináptica (LIEBERMAN, 2020).
Em modelos experimentais, identificou-se que a acidificação do autolisossomo diminui nos neurônios antes da deposição de Aβ extracelular em autolisossomos desacidificados aumentados. Em neurônios mais comprometidos, profusos vacúolos autofágicos (AVs) para Aβ se acumulam em grandes bolhas de membrana formando rosetas pericárias semelhantes a flores. Este padrão único, denominado PANTHOS (venenoso), também está presente em cérebros com DA. Os AVs adicionais coalescem em redes perinucleares de túbulos de membrana onde o Aβ se acumula intraluminalmente. com a permeabilização da membrana lisossômica, liberação de catepsina e morte celular lisossômica, acompanhada de invasão microglial. Análises quantitativas confirmam que neurônios individuais que exibem “Panthos” são a principal fonte de placas senis em modelos de APP.
A autofagia abrange vários mecanismos para sequestrar substratos e sua entrega aos lisossomos. Na principal via autofagia-lisossomal (ALP), a macroautofagia, uma membrana dupla alongada envolve o citoplasma ou, via proteína adaptadora, envolve substratos específicos e depois se fecha para formar um autofagossomo (AP) e amadurecem em autolisossomos (ALs) após fusão com lisossomo ou endolisossomo que introduz proteases de catepsina variadas, hidrolases ácidas e vATPase, a bomba de prótons que acidifica os lúmens de AL e ativa as hidrolases. Os LYs são alvos de produtos genéticos causadores e fatores de risco para AD5, incluindo os metabólitos patogênicos da APP (APP-βCTF e Aβ6) que são gerados ativamente nas vias endossomais e autofágicas e normalmente são eliminados pelos lisossomos (NIXON, 2007).
A acidificação dos autolisossomos diminui nos neurônios bem antes da deposição das placas Aβ associada à atividade da enzima ATPase marcadamente reduzida e ao acúmulo do fragmento carboxi-terminal da proteína precursora de amilóide nos autolisossomos desacidificados (LEE, 2022) apud MOURA, 2024a).
De acordo com Lee (2022), os AVs contendo substratos de autofagia incompletamente digeridos acumulam-se progressivamente nos neurônios afetados no estágio inicial da doença. A base molecular para a disfunção autofágica na DA, a sua relação com a patologia APP/amilóide e as suas implicações não são claras devido, em parte, aos desafios técnicos de monitorização de anomalias de ALP in vivo no cérebro.
Notavelmente, a fusão AV com o retículo endoplasmático (ER) produz a formação intraluminal de fibrillas beta-amilóide em uma rede tubular que circunda o núcleo, produzindo características morfológicas de uma placa amilóide central dentro do neurônio intacto. Nas técnicas de imagem e histoquímica, evidenciam-se que os neurônios “Panthos” são a origem da maioria das placas senis em modelos experimentais de DA, levando assim a uma reconsideração da sequência de eventos convencionalmente aceita na formação das placas Aβ na DA.
Nos modelos experimentais de DA, demonstrou-se deficiências precoces da atividade lisossômica, disfunção autofágica em populações de neurônios vulneráveis e acúmulo de Aβ dentro de AL pouco acidificado bem antes da deposição extracelular de Aβ. Além disso, identificou-se uma resposta autofágica única ao estresse em neurônios mais comprometidos, caracterizada pela proliferação fulminante de AVs dentro do pericário e formação de grandes bolhas de membrana repletas de AVs preenchidos com Aβ (LEE, 2022).
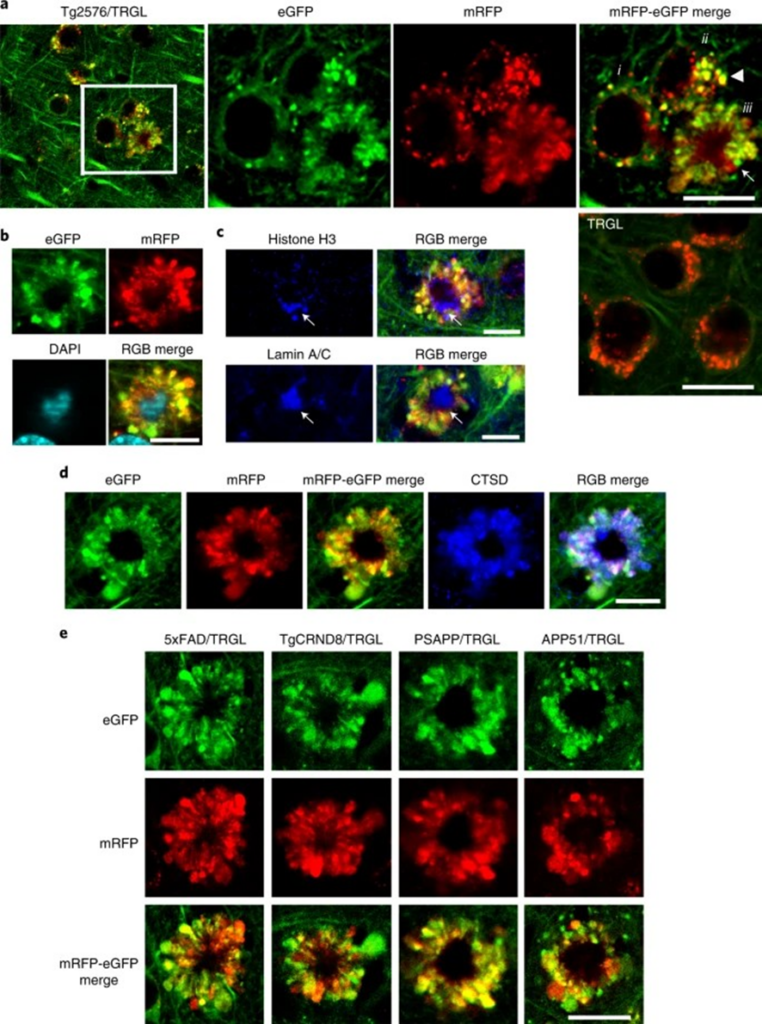
Fonte: Lee, 2022.
Sonda tfLC3 revela um padrão único de estresse autofágico, déficit de pH AL e formação de bolhas na membrana plasmática (‘PANTHOS’)
Lee (2022), observou um padrão neurodegenerativo autofágico idêntico em cinco modelos diferentes de camundongos com DA, incluindo modelos com início de neuropatologia acelerado (5xFAD, TgCRND8 e PSAPP) ou início tardio (Tg2576 e APP51 – um modelo de início excepcionalmente tardio que expressa hAPPwt)23. Camundongos 5xFAD/TRGL desenvolvem ruptura robusta de ALP e degeneração neuronal em idade precoce (começando após 2 meses, dependendo do sexo).
O autor utilizou este modelo em investigações adicionais sobre a relação entre o desenvolvimento de bolhas de membrana positivas para LC3 e a progressão da doença, incluindo patologia quantitativa da placa amilóide. Até onde sabemos, enormes saliências semelhantes da membrana pericária preenchidas por AV, conforme definido ultraestruturalmente, não foram descritas anteriormente em um estado neurodegenerativo . Como essas rosetas de grandes bolhas fluorescentes ao redor de um núcleo central positivo para DAPI se assemelham a pétalas de uma flor, denominamos esse processo degenerativo único de PANTHOS e nos referimos às células afetadas como neurônios PANTHOS.
A subsequente invasão microglial e astrocítica do neurônio PANTHOS anuncia a eventual morte celular que converte essa lesão amilóide dentro de um neurônio intacto em uma placa amilóide extracelular. A formação de placas β-amilóides na DA tem sido comumente considerada originária da deposição extracelular de β-amilóide derivada do Aβ secretado, que então desencadeia distrofia neurítica secundária e morte celular neuronal.
Por outro lado, nossas evidências em diversos modelos de DA apoiam a sequência oposta – ou seja, as placas extracelulares evoluem principalmente a partir do acúmulo intraneuronal de β-amilóide dentro dos túbulos da membrana, formando um ‘núcleo’ amilóide centralizado dentro de neurônios PANTHOS únicos e intactos que subsequentemente degeneram para dar ascender à placa senil clássica. Este processo ‘de dentro para fora’ concorda e fundamenta hipóteses de muitos investigadores. Nas versões desta hipótese, Aβ e suas espécies oligoméricas geradas intracelularmente dentro dos compartimentos de ALP podem obter acesso ao espaço extracelular por neurodegeneração, dano local à membrana ou secreção não convencional (exocitose). É importante ressaltar que alguns investigadores descreveram fibrilas amilóides envolvidas por membrana intracelular em modelos de camundongos com DA e, no cérebro com DA, a presença frequente de amilóide circundando núcleos positivos para DAPI e abundância de hidrolase lisossomal neuronal dentro de β-amilóide extracelular.
As descobertas acrescentam evidências crescentes de que a acidificação lisossomal e a desregulação do complexo vATPase são alvos comuns de perturbações genéticas e metabólicas associadas a doenças neurodegenerativas50. Juntamente com evidências anteriores, nossos achados apoiam fortemente uma ligação patogênica entre os metabólitos da APP e a disfunção do LY na DA. Notavelmente, a correção dos déficits de pH lisossômico relacionados ao PSEN1 por vários meios melhora a falha da autofagia e outras patologias relacionadas a DA.
Evidências demonstraram que a cascata PANTHOS em modelos de DA baseados em APP pode ser significativamente aliviada pelo direcionamento farmacológico do déficit de pH lisossômico. Além do significado dos achados aqui revelados, antecipamos o amplo potencial de nossa sonda transgênica de autofagia tfLC3 de dupla fluorescência para caracterizar as mudanças de ALP de forma sensível ao longo do tempo em outros modelos de doenças neurodegenerativas e para facilitar a avaliação de moduladores de autofagia / lisossoma como agentes terapêuticos (LEE, 2022).
A autofagia também é necessária para a plasticidade sináptica, ou seja, características sinápticas que mudam em estrutura, número e função para fortalecer ou enfraquecer o contato. Acredita-se que a plasticidade sináptica seja essencial para funções cognitivas como aprendizagem e memória. No nível celular, existem duas formas de plasticidade sináptica relacionadas a potenciação de longo prazo (LTP) e depressão de longo prazo (LTD).
Inicialmente, é relatado que a deficiência de BDNF (fator neurotrófico derivado do cérebro) pode regular e promover o acúmulo de autofagossoma, mas comprometer a LTP; em contraste, a LTP prejudicada devido à ablação do BDNF pode ser recuperada após a supressão da autofagia (NIKOLETOPOULOU et al., 2017), sugerindo que a indução da autofagia prejudica a LTP. No entanto, um estudo recente demonstrou que a estimulação da autofagia no hipocampo de camundongos era necessária para a formação de novas memórias e a LTP foi bloqueada após a inibição farmacológica da autofagia (GLATIGNY et al., 2019).
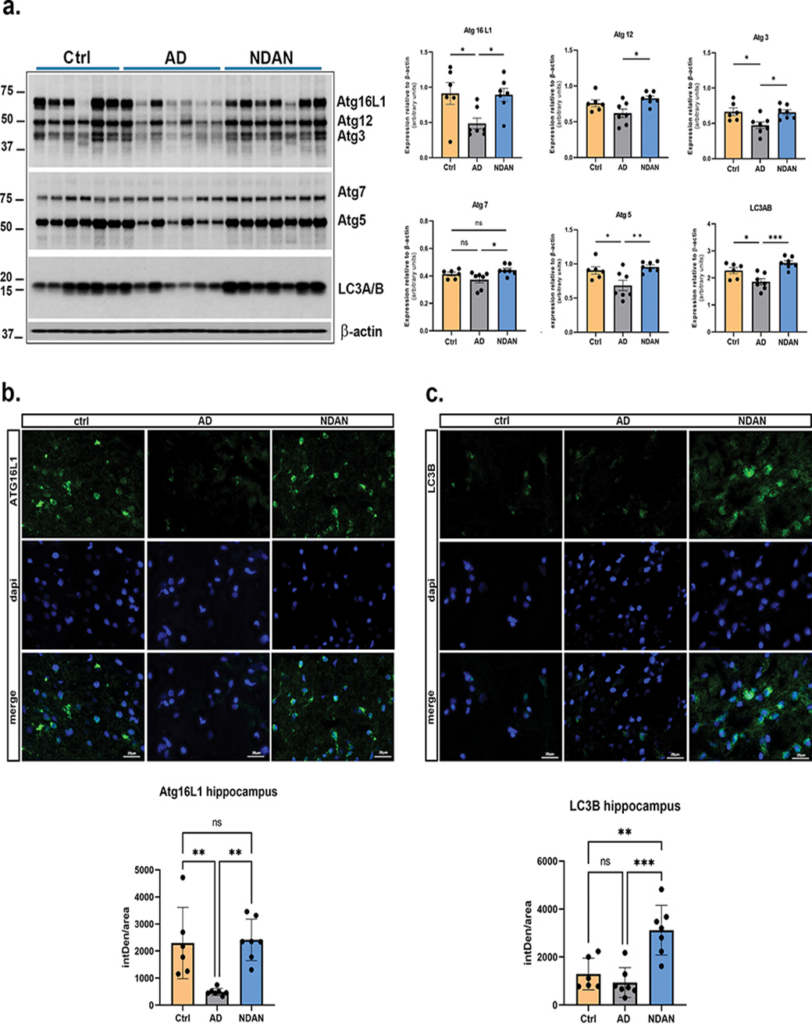
Fonte: Tumurbaatar, 2023.
Expressão e distribuição de proteínas de autofagia no hipocampo na DA.
Os resultados confirmam que certos indivíduos são mais responsivos do que outros a regulação da transcrição do gene da autofagia na presença de insultos patológicos da DA. É, portanto, prudente propor que tal capacidade de resposta aumentada contribua para a resistência observada contra o declínio cognitivo através da eliminação imediata de proteínas tóxicas relacionadas com a DA, particularmente a Tau. Pela primeira vez, a autofagia é preservada como um mecanismo protetor chave para a integridade cognitiva em indivíduos que permanecem resilientes ao declínio cognitivo apesar da neuropatologia da Tau (TUMURBAATAR, 2023).
Os mecanismos moleculares e as funções biológicas da macroautofagia e da autofagia foram extensivamente estudados, mas a microautofagia tem recebido muito menos atenção. Nos últimos anos, tem havido um crescimento nas pesquisas sobre microautofagia, primeiro em leveduras e depois em células de mamíferos. Neste estudo, os autores discutiram mecanismos regulatórios a montante, o crosstalk entre macroautofagia e microautofagia e as implicações funcionais da microautofagia em doenças como câncer e distúrbios neurodegenerativos em humanos. Por fim, os autores sugerem pesquisas futuras sobre microautofagia para desenvolver novas estratégias intervencionistas para doenças relacionadas à autofagia e aos lisossomos ((WAN et al, 2023).
MITOFAGIA
As deficiências na mitofagia no processo de degradação mitocondrial seletiva por autofagia leva a um acúmulo gradual de mitocôndrias defeituosas. A homeostase fisiológica das mitocôndrias é um equilíbrio entre a biogênese das mitocôndrias funcionais e a degradação autofágica das disfuncionais ou supérfluas por meio de um processo seletivo denominado mitofagia. Durante essa via de limpeza, as mitocôndrias disfuncionais são engolfadas por uma membrana dupla (fagóforo) formando o mitofagossomo que então se fundirá com um lisossomo para formar um mitofagolisossomo. Nessa estrutura, as hidrolases lisossomais digerirão as mitocôndrias em pequenos componentes que podem então ser reciclados.
Estudos identificaram dois tipos principais de mitofagia (PINK1/Parkin-dependente ou independente) ativadas por uma infinidade de estímulos e implicando diferentes proteínas que exibem um papel de receptores de mitofagia. Como os neurônios são locais de processos energéticos de alta demanda, sua homeostase é altamente dependente da saúde mitocondrial e da eficácia da mitofagia. Os neurônios apresentam a especificidade de que os lisossomos degradativos estão predominantemente localizados no soma, exigindo, portanto, que as mitocôndrias/mitofagossomos distais danificados sejam transportados de forma retrógrada de volta ao corpo celular para sua degradação por meio de maquinário de transporte. O controle de qualidade mitocondrial axonal também pode ocorrer de forma dependente ou independente de Parkin, proporcionando, assim, uma neuroproteção rápida contra condições de estresse ou estágios iniciais de doenças neurodegenerativas (LIN, 2017; ASHRAFI, 2014 apud MARY, 2023).
O comprometimento mitocondrial ocorre antes do estágio patológico associado a DA e, portanto, pode atuar como um “fator de risco”. O exame dos resultados em modelos animais e nos cérebros de pacientes com DA, levou a hipótese de que a mitofagia prejudicada é um aspecto importante da doença e pode estar tanto a montante quanto a jusante de Aβ e Tau em um ciclo vicioso, causando, em última análise, disfunções sinápticas e déficits cognitivos (KERR, 2019).
A redução da mitofagia foi caracterizada pelo acúmulo de mitocôndrias estrutural e funcionalmente danificadas (tamanho reduzido, cristas desorganizadas e baixa produção de ATP) e por um comprometimento das etapas de iniciação do processo de mitofagia (recrutamento reduzido de LC3 ativado para mitocôndrias e uma cascata disfuncional de proteína quinase ativada por AMP (AMPK), caracterizada pela fosforilação de AMPK e pela inibição de seus alvos (ULK1 e TBK1). Consequentemente, essas observações foram consolidadas por uma fusão defeituosa de mitofagossomos com lisossomos e um acúmulo de mitocôndrias danificadas em AVs em cérebros com DA (YE, 2015; FANG, 2019).
O transporte retrógrado de mitocôndrias distalmente danificadas em direção ao soma do neurônio, onde elas deveriam ser degradadas nos mitolisossomos, também é prejudicada em pacientes com DA. A atividade reduzida dos complexos mitocondriais I, II e V ocorre já nos estágios I-II da DA no córtex entorrinal (CE), sugerindo que as disfunções mitocondriais e a falha da mitofagia ocorrem principalmente na região cerebral vulnerável. Essas alterações provavelmente são amplificadas por Aβ e contribuem para o acúmulo de Tau durante os estágios prodrômicos da DA (KOBRO-FLATMOEN, 2021).
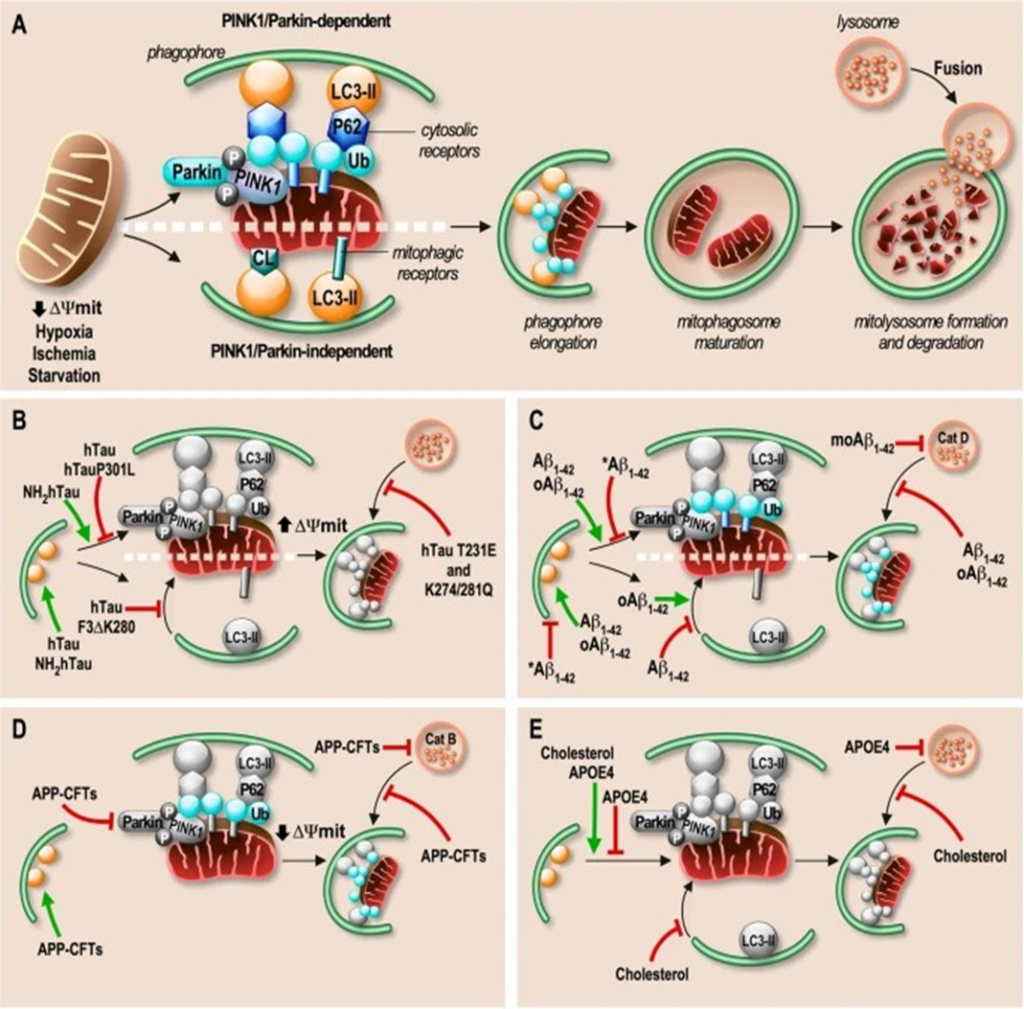
Mary, 2023.
O acúmulo de evidências baseadas em modelos genéticos, moleculares e pré-clínicos apóia o direcionamento da mitofagia em doenças neurodegenerativas. Apesar dos desafios de desenvolvimento clínico, abordagens baseadas em pequenas moléculas para o aprimoramento seletivo da mitofagia – inibidores USP30 e ativadores PINK1 – estão entrando em ensaios clínicos de fase I (ANTICO, 2025).
Chaplygina (2025) avaliou a importância dos diferentes tipos de fenótipos mitocondriais no tecido nervoso, discutindo como as mitocôndrias na DA ficam “presas” em determinados padrões e como esse padrão se mantém. Compreender os papéis específicos e as transições entre as formas mitocondriais, incluindo minúsculas, em rede e hiperfundidas, é crucial no desenvolvimento de novas terapias destinadas a restaurar a homeostase mitocondrial. Ao visar estas dinâmicas, é possível intervir precocemente no processo da doença, oferecendo novos caminhos para prevenir ou tratar a neurodegeneração.
METABOLISMO MITOCONDRIAL
A disfunção mitocondrial impulsiona a patogênese da DA, uma vez que a função basal e as taxas de alteração influenciam a progressão do declínio cognitivo. Além disso, os neurônios apresentam a especificidade de que os lisossomos degradativos estão predominantemente localizados no soma, exigindo, portanto, que as mitocôndrias/mitofagossomos distais danificados sejam transportados de forma retrógrada de volta ao corpo celular para sua degradação. O controle de qualidade mitocondrial axonal também pode ocorrer de forma dependente ou independente de Parkin, proporcionando, assim, uma neuroproteção rápida contra condições de estresse ou estágios iniciais de doenças neurodegenerativas (LIN, 2017; ASHRAFI, 2014 apud MARY, 2023).
A mitofagia refere-se à despolarização das mitocôndrias durante o estresse oxidativo, o envelhecimento e os danos as ERO, e pode executar o encapsulamento das mitocôndrias danificadas no autofagossomo, onde se funde com os lisossomos intracelulares para a degradação das mitocôndrias danificadas para manter a homeostase celular (FANG, 2019).
A mitofagia foi identificada como uma forma de macroautofagia que resulta na degradação seletiva de mitocôndrias disfuncionais ou danificadas e desempenha um papel vital na manutenção da função celular, reduzindo o estresse oxidativo e restaurando a homeostase celular (PRADEEPKIRAN, 2020).
Evidências recentes sugerem que o estresse oxidativo também causa disfunção mitocondrial e está associado ao desenvolvimento da DA (YU et al., 2018). Impulsionados pela energia, os neurônios estabelecem o potencial de membrana e sintetizam, secretam e reciclam neurotransmissores, mantendo assim a homeostase do Ca2+ intracelular e transmitem efetivamente os sinais neuronais. Estudos atuais demonstraram que a cadeia respiratória mitocondrial e as enzimas do ciclo do TCA são significativamente reduzidas na DA (BUTTERFIELD et al., 2006).
Da mesma forma, a cadeia respiratória mitocondrial prejudicada irá desordenar ainda mais a eficiência da transferência de elétrons, aumentar o conteúdo de ERO e levar a lesões celulares. Além disso, o DNA mitocondrial (mtDNA) localizado próximo à cadeia respiratória mitocondrial é suscetível as mutações resultando em proteínas defeituosas e ERO (GRIMM et al., 2016).
Neste contexto, o estresse oxidativo é uma das marcas mais proeminentes do processo de envelhecimento, caracterizado pela combinação do sistema de defesa antioxidante reduzido e da atividade prejudicada da fosforilação oxidativa mitocondrial (OXPHOS) (GRIMM, 2017).
Os neurônios que dependem do OXPHOS para atender às suas demandas energéticas são propensos ao hipometabolismo energético. Da mesma forma, devido às características de não divisão, os neurônios não poderiam ser substituídos, exceto que o hipocampo gere continuamente novos neurônios durante o período da idade adulta (SANCHEZ-VARO et al., 2012).
As evidências confirmam que a falha energética e o estresse oxidativo causado pela disfunção respiratória mitocondrial desempenham um papel crucial na patogênese da DA (MARQUES-ALEIXO et al., 2015).
Devido à ausência de ação da telomerase em muitas células somáticas, o comprimento do telômero vai encurtando a cada divisão celular, até a célula entrar em senescência. Diversos estudos demonstraram que o envelhecimento celular está associado à redução da integridade funcional das mitocôndrias e, consequentemente, ao aumento da produção de radicais livres e ERO. Alguns autores da teoria mitocondrial do envelhecimento sugerem que mutações ocorridas no genoma mitocondrial alteram o metabolismo mitocondrial, reduzindo a produção de ATP e predispondo a célula ao envelhecimento e a diversas doenças (MARY, 2023).
Além de serem os provedores de energia e metabólitos, as mitocôndrias geram espécies reativas de oxigênio (ROSmit), que atuam como sensores fisiopatológicos de uma infinidade de eventos celulares, incluindo morte celular, proliferação, diferenciação e imunidade. A homeostase fisiológica das mitocôndrias é o equilíbrio entre a biogênese das mitocôndrias funcionais e a degradação autofágica das disfuncionais ou supérfluas por meio de um processo denominado mitofagia. Durante essa via de limpeza, mitocôndrias são engolfadas por uma membrana dupla (fagóforo) formando o mitofagossomo, que então se fundirá com um lisossomo para formar um mitofagolisossomo. Nessa estrutura, as hidrolases lisossomais digerem as mitocôndrias em pequenos componentes que podem então ser reciclados FERREIRA, 2022).
Martín-Maestro et al. (2024) observaram em cérebros afetados pela DA uma regulação negativa da expressão de várias proteínas conhecidas por participarem dos processos de autofagia e mitofagia, observados nos cérebros de pacientes heterozigotos do gene APOEε4.
Fang et al. (2019), mostraram que os níveis basais de mitofagia são reduzidos em 30 a 50% em amostras de cérebro hipocampal post-mortem de pacientes com DA em comparação com pacientes sem alterações cognitivas pareados por sexo e idade. A redução da mitofagia foi caracterizada pelo acúmulo de mitocôndrias estrutural e funcionalmente danificadas (tamanho reduzido, cristas desorganizadas e baixa produção de ATP) e por um comprometimento das etapas de iniciação do processo de mitofagia (recrutamento reduzido de LC3 ativado para mitocôndrias e uma cascata disfuncional de proteína quinase ativada por AMP (AMPK), caracterizada pela fosforilação de AMPK e pela inibição de seus alvos moleculares. Consequentemente, essas observações foram consolidadas por uma fusão defeituosa de mitofagossomos com lisossomos e um acúmulo de mitocôndrias danificadas em AVs em cérebros com DA.
Wang et al (2024) relataram que DISC1, uma proteína que regula o tráfego de mitocôndrias axonais, é menos abundante no córtex pré-frontal. Surpreendentemente, os autores também demonstraram que a DISC1 age como um receptor de mitofagia presente no OMM recrutando fagóforos para mitocôndrias danificadas. A atividade reduzida dos complexos mitocondriais I, II e V ocorre na DA no córtex entorrinal (CE) sugerindo que as disfunções mitocondriais e a falha da mitofagia está localizada principalmente na região cerebral vulnerável. Essas alterações provavelmente são amplificadas por Aβ e contribuem para o acúmulo de tau durante os estágios prodrômicos da DA.
Cummings et al. (2024) descobriram que a superexpressão de Tau (uma mutação associada à demência frontotemporal) inibiu a mitofagia em células de neuroblastoma. Os autores notaram que o bloqueio da mitofagia é uma interação aberrante entre o domínio de projeção da proteína Tau e Parkin, prejudicando sua translocação para mitocôndrias danificadas. Os resultados demonstraram que o comprometimento da mitofagia pode estar associado à patogênese da Tau e ressaltaram que conclusões distintas podem ser tiradas de acordo com a natureza da proteína Tau analisada e com o modelo e os métodos usados para monitorar a mitofagia.
A disfunção mitocondrial desempenha um papel principal na progressão de um amplo espectro de doenças degenerativas, incluindo, entre outras, insuficiência cardíaca, hipertensão, obesidade, diabetes, insuficiência hepática, insuficiência renal e DA. A qualidade mitocondrial é ajustada por uma miríade de sistemas interconectados, incluindo (1) elementos enzimáticos e não enzimáticos capazes de combater a toxicidade mitocondrial mediada pelo oxigênio; (2) proteases e acompanhantes mitocondriais responsáveis pela manutenção da proteostase mitocondrial; e (3) uma rede multicamadas de proteínas envolvidas no controle da morfologia, localização e número mitocondrial (FERREIRA, 2022).
Fonte: Ferreira, 2022.
Mitocôndria envolvida pelo autofagossoma
No modelo experimental de Tostes et al (2022), os autores utilizaram a segregação de mtDNA usando uma linhagem de camundongos heteroplasmáticos bem estabelecida com mtDNA de origem NZB / BINJ e C57BL / 6N. Esta linhagem de camundongos mostrou um acúmulo pronunciado de mtDNA dependente da idade no fígado, levando assim a uma maior capacidade respiratória por molécula de mtDNA. Notavelmente, o nocaute Atg7 específico do fígado (relacionado à autofagia) aboliu o acúmulo de mtDNA resultando em segregação de mtDNA quase neutra durante o desenvolvimento até a idade adulta. Assim, os pesquisadores propõem que a segregação do mtDNA hepático relacionada à idade é uma consequência da depuração macroautofágica do mtDNA.
Guglielmotto et al (2024), relataram que o Aβ desencadeia um bloqueio do fluxo de autofagia em células diferenciadas de neuroblastoma humano. Isso foi evidenciado pelo acúmulo de P62 e de autofagossomos e pela atividade lisossomal reduzida Dados emergentes sugerem que o acúmulo precoce de C99, em vez de Aβ, se correlaciona com defeitos de autofagia e mitofagia na DA. Consequentemente, o acúmulo de C99 ocorreu em vários modelos de camundongos transgênicos nos modelos experimentais de DA. É importante ressaltar que um estudo recente relatou que o acúmulo de C99 se correlaciona com a vulnerabilidade neuronal em cérebros humanos afetados pela DA.
Os resultados do estudo supramencionado aponta alteração da estrutura da mitocôndria na produção de ROSmit e nos fenótipos de falha da mitofagia exacerbados pela inibição da γ-secretase que bloqueia a produção de Aβ e potencializa o acúmulo de APP-CTFs (C99 e C83). A inibição da β-secretase (que previne a produção de Aβ e C99) tende a melhorar a estrutura da mitocôndria e a resgatar o defeito da atividade do complexo I. Confirmaram ainda o papel dos APP-CTFs na superprodução de ROSmit e a falha da mitofagia expressando apenas o fragmento C99. A inibição farmacológica da γ-secretase em AD-iNSC piorou as disfunções mitocondriais e de mitofagia. No geral, esses resultados demonstraram firmemente a contribuição de APP-CTFs, independentemente de Aβ, para defeitos de mitofagia.
Portanto, o acúmulo de mitocôndrias danificadas poderia resultar tanto em alterações neuropatológicas quanto em sintomas da DA. Nesta hipótese, o envelhecimento representa um importante fator de risco para o desenvolvimento da DA esporádica e o acúmulo de Aβ é o resultado induzido pelo envelhecimento, e não a causa da evolução neuropatológica (SWERDLOW et al., 2018).
A hipótese da cascata mitocondrial é um complemento à hipótese da cascata amilóide, sugerindo o importante papel do estado funcional das mitocôndrias na produção, modificação e acumulação de Aβ e Tau, bem como na formação de oligômeros (SWERDLOW, 2018). Portanto, a disfunção mitocondrial pode ser um indutor a montante de Aβ e Tau hiperfosforilada, enquanto Aβ e Tau hiperfosforilada podem exacerbar ainda mais a disfunção mitocondrial.
IMUNOMETABOLISMO
Desde 1906, o caminho para a cura da DA tem sido pavimentado por inúmeras hipóteses descritivas e sucessivos fracassos de ensaios clínicos. Por outro lado, a era dos estudos de associação genômica ampla revolucionou a visão “neurocêntrica” clássica da DA, fornecendo pistas de que as células imunológicas residentes no cérebro (micróglia e astrócitos) também são “atores-chave” na trajetória patológica e clínica desse distúrbio neurodegenerativo. Considerando que a intercomunicação entre neurônios, astrócitos e micróglia é fundamental para a organização funcional do cérebro, fica evidente que a interrupção do bom funcionamento desta “tríade” poderia contribuir para os eventos neuroinflamatórios e neurodegenerativos que ocorrem no cérebro com DA.
É importante ressaltar que os recentes progressos científicos no campo do imunometabolismo – interface entre o metabolismo e a resposta imunitária – lançam luz sobre a importância da reprogramação metabólica das células imunitárias residentes no cérebro na DA. Neste sentido este capítulo tem como objetivo discutir o conhecimento atual sobre os padrões metabólicos das células imunitárias residentes no cérebro durante o continuum da DA e patologias relacionadas, endofenótipo imunometabólico bem como apresentar o novo modelo de DA como Doença Autoimune (AD2).
Imunometabolismo é a comunicação e interdependência entre o sistema imunológico e o metabolismo celular. As interações em múltiplos níveis entre os sistemas metabólico e imune sugerem que mecanismos patogênicos podem estar subjacentes às complicações observadas assevera Moraes-Vieira (2016).
A inflamação crônica devido aos distúrbios metabólicos pode se propagar para o SNC pela entrada de moléculas inflamatórias na área do tronco cerebral, o que pode afetar os núcleos colinérgicos como núcleo tegmental, núcleo septal medial, banda diagonal de Broca e núcleo basal magnocelular. Há também evidências de que a inflamação periférica devido à obesidade precipita a inflamação cerebral. Os resultados sugerem que a inflamação periférica e central estão interrelacionadas e implicadas no comprometimento cognitivo devido a disfunção colinérgica que ocorre em pacientes com distúrbios metabólicos (TANAKA, 2020).
Por ser uma doença multifatorial, evidências mostram que estes eventos estão relacionados a alterações adicionais no cérebro de pacientes com DA, como a neuroinflamação, o estresse oxidativo e a perda sináptica dos neurônios colinérgicos. Ao longo dos anos, diversas hipóteses foram estabelecidas a fim de obter um melhor entendimento dos mecanismos envolvidos na neuropatologia da DA (LIU et al., 2019).
O cérebro também pode afetar o sistema imunológico ao liberar antígenos, citocinas e quimiocinas derivados do cérebro por meio de infiltração, ativação e polarização de células imunológicas no SNC (MUHAMMAD et al., 2021). Um crescente conjunto de evidências sugere a presença de características semelhantes à memória nas células do sistema imunológico inato. Certas infecções, vacinações ou moléculas podem reprogramar células imunes inatas para exibir uma resposta mais forte a lesões secundárias, um fenômeno conhecido como imunidade treinada mediada por uma ampla gama de alterações epigenéticas e metabólicas (BULUT et al., 2021).
Em pacientes com DA, as células imunológicas sofreram várias alterações metabólicas, como glicose desregulada, metabolismo lipídico, funções mitocondriais e imunidade inata comprometida no cérebro, resultando em maior suscetibilidade a infecções e patologia exacerbada da DA (SHINJYO, 2021; WU et al, 2021).
Na DA, a atividade neuronal poderia modular a liberação da proteína tau. Uma vez liberada a proteína tau, ela poderia ser internalizada por outros neurônios e, por meio de interações com proteínas tau nos neurônios receptores, desencadear um dobramento anormal, avançando assim a progressão da doença (PEREA et al., 2020; UEMURA et al., 2020).
A transmissão da proteína tau não se limitou apenas aos neurônios, mas também esteve envolvida na fagocitose microglial, onde a microglia engolfou a proteína tau ou os neurônios que transportam a proteína tau, resultando em sua ativação imunológica e subsequente morte neuronal (BRELSTAFF et al., 2018; GIBBONS et al, 2019).
A micróglia ativada pela proteína Tau pode levar a irregularidades metabólicas, incluindo o acúmulo anormal de succinato e a interrupção do ciclo do TCA. Estes fenómenos contribuíram ainda mais para o desenvolvimento da neuroinflamação (ZHAO et al., 2022).
Como núcleo do metabolismo das células imunológicas, as mitocôndrias podem influenciar diretamente o destino e a aptidão celular através da regulação do desenvolvimento, ativação, proliferação, diferenciação e morte celular. Além disso, as ERRO produzidas pelas mitocôndrias podem afetar a resposta imune celular durante a atividade fisiológica. As ERO como subprodutos da cadeia de transporte de elétrons (ETC) estavam intimamente associadas ao dano oxidativo, mas também poderiam atuar como uma importante molécula sinalizadora para a ativação celular (CHEN, 2023).
Comorbidades como diabetes mellitus, obesidade, hipertensão, doenças cerebrovasculares e cardiovasculares, dislipidemia, depressão, sarcopenia, resistência à insulina, má nutrição, precursores de anorexia do envelhecimento e distúrbios hormonais incluindo hipogonadismo e hipovitaminose D também desempenham papéis na etiologia da DA (BESTEN et al., 2013; JENA et al., 2018; KANDIMALA et al., 2016; SCHMIDT, 2018, apud TICINESI et al. 2018; SILVA et al., 2019).
A análise de correlação baseada na avaliação bioenergética em tempo real e PET com fluorodesoxiglicose (FDG) revela que a utilização e o metabolismo da glicose cerebral restaurados pelo tratamento com GAF NP podem servir como um indicador sensível e eficaz para a mudança de polarização microglial M1 para M2, em última análise, aliviando a neuroinflamação e sua neurodegeneração derivada, bem como melhora o declínio cognitivo em camundongos com DA. Este trabalho destaca uma potencial nanomedicina destinada a modificar a reprogramação imunometabólica mediada por mTOR para interromper a progressão da DA induzida pela privação de energia (YANG, 2023).
As síndromes metabólicas estão frequentemente associadas à demência, sugerindo que a desregulação do metabolismo energético pode aumentar o risco de neurodegeneração e comprometimento cognitivo. Além disso, evidências crescentes sugerem a ligação entre infecções e distúrbios cerebrais, incluindo a DA. O sistema imunológico e o metabolismo energético estão em uma relação intrincada. A infecção desencadeia respostas imunológicas, que são acompanhadas por desequilíbrio no metabolismo energético celular e do organismo, enquanto distúrbios metabólicos podem levar à desregulação imunológica e maior suscetibilidade à infecção. No cérebro, as atividades das células imunológicas residentes no cérebro, incluindo a micróglia, estão associadas às suas assinaturas metabólicas, que podem ser afetadas pela infecção do sistema nervoso central (SNC).
Evidências sugerem que distúrbios metabólicos podem causar distúrbios neurodegenerativos, incluindo a DA. Possivelmente por meio de ruptura da barreira hematoencefálica (BHE) e neuroinflamação. A associação também pode ser agravada e influenciada por outros fatores, como a idade indicando a complexidade dos mecanismos subjacentes à ligação entre distúrbios metabólicos e declínio cognitivo. Entretanto, a relação entre infeções e demência, especialmente DA de início tardio, tem sido repetidamente sugerida nas últimas três décadas (ASHRAF et al., 2019; MEJIDO et al., 2020; PUGAZHENTHI et al., 2020).
Agusti, et al.(2018), indicam que a obesidade está associada ao maior risco de desenvolvimento de depressão, disbiose, diabetes tipo 2 e DA onde as diferentes patologias podem envolver mecanismos compartilhados, sendo a microbiota intestinal um fator mediador que influencia a fisiologia e pode explicar o vínculo entre essas patologias. Portanto, a microbiota intestinal tem efeitos significativos na estrutura e função do sistema nervoso entérico e central, incluindo o comportamento e a regulação do cérebro.
O metabolismo celular e sistêmico afeta as respostas imunológicas. Para o autor, a exposição a proteína Aβ pode aumentar a glicólise, fagocitose e quimiotaxia, mas a exposição crônica causa desregulação metabólica e funções imunológicas comprometidas de acordo com os estudos advindos do imunometabolismo (SHIPPY, 2020). Dados observacionais e estudos intervencionistas sugerem um possível elo entre a ingestão de glúten e o desenvolvimento de transtornos neurológicos. A perda da integridade da barreira hematoencefálica (BBB), atribuída à inflamação, está sendo relacionada a várias condições neurológicas, sugerindo que a inflamação mediada pelo glúten pode desempenhar um papel na disfunção da BBB e contribuir para a neurodegeneração.
Phillip (2022) aponta uma correlação do sistema imunológico produzindo anticorpos relacionados ao glúten e a ocorrência de transtorno bipolar, depressão, ansiedade, mania, fobias sociais e hiperatividade sugerindo que uma dieta sem glúten poderia ser uma opção de tratamento plausível para melhorar os sintomas do transtorno de humor. Por outro lado, a transglutaminase é implicada na resposta inflamatória ao glúten, mediando a desaminação de proteínas do glúten. Ainda, a isoforma neural da transglutaminase está implicada na patogênese de várias doenças neurodegenerativas, incluindo a DA.
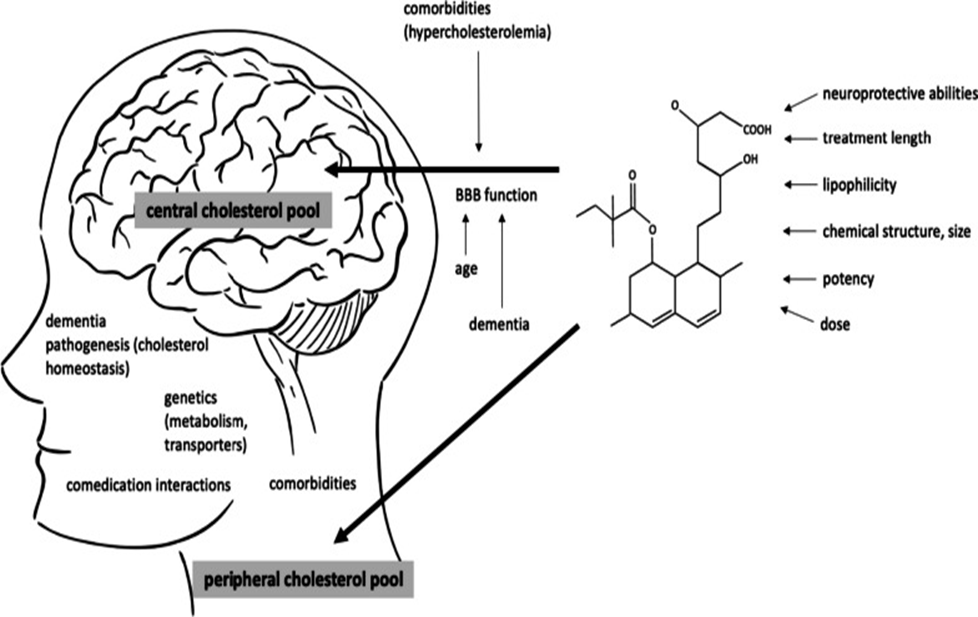
Fonte: Garcia-Ptacek, 2023.
Petek (2023) concluíram que inúmeros pacientes com DA ou demência mista com indicação de medicação hipolipemiante podem se beneficiar cognitivamente do tratamento com estatinas; no entanto, são necessárias mais pesquisas para esclarecer os resultados das análises de sensibilidade.
“DIABETES TIPO 3”
Pesquisadores propuseram o termo ‘Diabetes Tipo 3” para a DA devido às características moleculares e celulares compartilhadas entre Diabetes Tipo 2 e a resistência à insulina associada a déficits de memória e declínio cognitivo em idosos. Por exemplo, a insulina está envolvida na ativação do glicogênio sintase quinase 3β que por sua vez causa fosforilação de Tau, a qual está envolvida na formação de emaranhados neurofibrilares. Curiosamente, a insulina também desempenha um papel crucial na formação de placas Aβ (KANDIMALLA, 2016).
O Diabetes Tipo 3 (DT3) é um distúrbio neuroendócrino que representa a progressão do Diabetes Mellitus Tipo 2 (DM2) para a DA. A enzima degradadora de insulina (IDE) pode ser o principal ator que detém a capacidade de mudar o DM2 para o DM3, alterando as vias metabólicas, como a regulação do desenvolvimento de células β, a regulação negativa das vias PI3K/AKT e a degradação da Aβ (MITTAL, 2016).
Kandimalla (2017) assevera que durante muito tempo acreditava-se que a insulina não tinha ligação com o sistema nervoso central, mas nos anos, vários grupos de pesquisa localizaram o hormônio e seu receptor no cérebro. Pouco tempo depois foi descoberto que o hormônio desempenha papel importante no aprendizado e na memória. Atualmente tem surgido estudos indicando que o metabolismo da glicose e a resistência à insulina estão envolvidos com os eventos patológicos decorridos na DA. Além disso, foi relatado que pacientes diabéticos possuem a capacidade mental diminuída e possuem o dobro de chances de desenvolverem a DA. Devido a isso, alguns pesquisadores começaram a considerar a DA como “Diabetes tipo 3” (AHN et al., 2019).
A insulina possui um papel importante nos processos de formação de memória e aprendizagem, além de estar envolvida com a LTP (YANG et al., 2018). Em um estado de resistência à insulina ou de diminuição de IGF-1 ocorre o aumento da atividade da GSK-3β que possui um importante papel na hiperfosforilação da proteína Tau, podendo levar a formação dos emaranhados neurofibrilares presentes na DA (ARNOLD et al., 2018).
Segundo Westwood et al., (2024) a regulação do IGF-1R no cérebro causa a formação de oligômeros e comprometimento cognitivo, o que aumenta as chances de desenvolver a DA em pacientes diabéticos. A maior expressão de IGF-1R também contribui para o desenvolvimento da DA em idosos. Portanto, tanto a regulação positiva quanto a regulação negativa do IGF-1R afetam o cérebro. Embora o IGF-1R seja um alvo drogável, as terapias baseadas no fator de crescimento falharam nos ensaios clínicos. O IGF-1R pode ativar ainda mais as interleucinas e criar estresse oxidativo excessivo no tecido cerebral.
Maedler et al. (2024) relatou que sinais perturbados de IL1B estão envolvidos na progressão da resistência à insulina e levam ao DM2. Essas interleucinas ativadas e a alfa-2 formam um complexo e eventualmente regulam positivamente a expressão de APOE. Terapias como o anticorpo anti-APOE já estão em testes clínicos para diminuir a carga de amilóide no cérebro com DA.

Fonte: Cai, 2022
Os impactos combinados e multifatoriais devido a essas interações podem agravar ainda mais a disfunção cerebral. Como principais reguladores do metabolismo energético e das respostas imunológicas, a insulina e a leptina desempenham papéis significativos nessas intrincadas interações. Deve-se notar também que outros hormônios metabólicos e adipocinas, como o glucagon e a adiponectina, participam de forma semelhante no imunometabolismo, e a desregulação dessas moléculas também está implicada em distúrbios neurodegenerativos, incluindo a DA (GRIECO et al., 2019).
DISBIOSE
O trato gastrointestinal humano é povoado por uma comunidade microbiana diversificada. O vasto potencial genético e metabólico do microbioma intestinal sustenta a sua onipresença em quase todos os aspectos da biologia humana, incluindo a manutenção da saúde, o desenvolvimento, o envelhecimento e as doenças. O advento de novas tecnologias de sequenciamento e métodos independentes de cultura in vivo e em vitro permitiu aos pesquisadores ir além dos estudos correlativos em direção a explorações para lançar luz sobre as interações microbioma-hospedeiro. As evidências revelaram a comunicação bidirecional entre o microbioma intestinal e o sistema nervoso central, conhecida como “eixo microbiota-intestino-cérebro”. O eixo microbiota-intestino-cérebro representa um importante regulador das funções gliais, tornando-o um alvo acionável para melhorar o desenvolvimento e a progressão de doenças neurodegenerativas (LOH, 2024).
A microbiota intestinal interage continuamente com os sistemas nervoso, imunológico e endócrino para garantir o desenvolvimento e funcionamento adequados de cada sistema (PLUTA, 2023 apud LIAO, 2024).
A translocação de bactérias comensais, consequentemente, geram mais implicações. Evidências recentes mostraram que bactérias isoladas do intestino tem a capacidade de sintetizar compostos neuroativos incluindo neurotransmissores, muitos dos quais resultam do catabolismo de aminoácidos (PAREDE et al., 2014).
As bactérias intestinais desempenham um papel importante na digestão dos alimentos, na ativação imunológica e na regulação das vias de sinalização enteroendócrinas, mas também se comunicam com o SNC através da produção de compostos metabólicos específicos como ácidos biliares, ácidos graxos de cadeia longa (SCFAs), glutamato (Glu), ácido γ-aminobutírico (GABA), dopamina (DA), norepinefrina (NE), serotonina (5-HT) e histamina (DICKS, 2022).
De acordo com Dicks (2022), as fibras aferentes do nervo vago (VN) que transportam sinais do trato gastrointestinal (TGI) e da microbiota intestinal para o cérebro também estão ligadas a receptores no esôfago, fígado e pâncreas. Em resposta a estes estímulos, o cérebro envia sinais às células enteroepiteliais através do VN. Os sinais chegam à microbiota intestinal através de 100 a 500 milhões de neurônios do SNE na submucosa e no plexo mioentérico da parede intestinal.
A modulação, o desenvolvimento e a renovação dos neurônios do SNE são controlados pela microbiota intestinal, especialmente aqueles com capacidade de produzir e metabolizar hormônios. Os sinais gerados pelo hipotálamo alcançam as glândulas pituitária e adrenal e se comunicam com as células enteroepiteliais através do eixo hipotálamo-hipófise-adrenal (HPA). Os SCFAs produzidos pelas bactérias intestinais aderem aos receptores de ácidos graxos livres (FFARs) na superfície das células epiteliais intestinais (IECs) e interagem com os neurônios ou entram no sistema circulatório. Além disso, as bactérias intestinais alteram a síntese e a degradação dos neurotransmissores (DICKS, 2022).
A disfunção do eixo intestino-cérebro, bem como a presença de anticorpos para a decarboxilase do ácido glutâmico (GAD) sugerem vias alternativas através das quais o glúten pode influenciar a função neuronal. Embora as evidências diretas que ligam o consumo de glúten à neurodegeneração sejam limitadas, a relação entre o glúten, a produção de anticorpos e a disrupção do eixo microbiota-intestino-cérebro fornece insights importantes (PHILIP, 2022).
Os microrganismos influenciam as vias neurais regulando a circulação de fatores imunológicos e metabólitos, que, por sua vez, afetam o curso das doenças neurodegenerativas (CARABOTTI et al., 2015). O eixo microbioma-intestino-cérebro refere-se à resposta funcional dos micróbios intestinais ao eixo intestino-cérebro em humanos (LIU et al., 2022; SANTOS et al., 2023).
A disbiose pode colaborar para o desenvolvimento da DA, principalmente, em relação a ativação de citocinas pró-inflamatórias, levando ao aumento da permeabilidade intestinal e ao desenvolvimento da resistência insulínica. Estudos afirmam que há uma comunicação entre o trato gastrointestinal, a microbiota e o sistema nervoso central, possuindo assim, uma relação harmoniosa no qual todos exercem funções e sofrem influência dos mesmos. Assim, quando há um desequilíbrio em algum dos eixos, há consequências que podem levar ao desenvolvimento de doenças. Existem evidências crescentes que afirmam a alteração da microbiota intestinal ocasionando o desequilíbrio do eixo cérebro-intestino traduzindo-se em transtornos do humor podendo evoluir para DA (SILVESTRE, 2016).
O conceito de eixo intestino-cérebro foi estendido para eixo microbiota-intestino-cérebro, visto que as bactérias presentes no lúmen intestinal exercem papel fundamental na comunicação com o SNC. A estabilidade da microbiota tende a acontecer na idade adulta, todavia, caso ocorra a disbiose, ainda existe a possibilidade de afetar a função e o comportamento cerebral, visto que a poda sináptica e a mielinização não deixam de ocorrer nesse estágio. Durante o envelhecimento, há um declínio gradual da harmonia da microbiota, isso ocorre devido as respostas pró-inflamatórias progressivas e crônicas, que podem ser acompanhadas pela redução do peso cerebral e das funções cognitivas (LANDEIRO, 2016; PISTOLATTO, 2016).
NERVO VAGO E MICROBIOTA
A microbiota interage com o cérebro através de várias vias e processos, incluindo metabólitos, nervo vago, eixo HPA, sistema endócrino e sistema imunológico para manter a homeostase cerebral. A disbiose está associada a vários distúrbios neurológicos, incluindo ansiedade, depressão e DA. Por outro lado, a DA é sustentada por neuroinflamação e neurodegeneração mediadas por micróglia. Além disso, os GM e seus produtos também afetam a neuroinflamação e a neurodegeneração mediadas pela micróglia. Apesar das evidências que conectam a disbiose com progressão da DA, o envolvimento do GM na modulação da neuroinflamação mediada pela micróglia na DA permanece indefinido. É importante ressaltar que decifrar o(s) mecanismo(s) pelos quais o GM regula a neuroinflamação dependente da micróglia pode ser útil no desenvolvimento de possíveis estratégias terapêuticas para mitigar a DA (BANO, 2024).
Vários grupos taxonômicos de bactérias podem sintetizar neurotransmissores, como dopamina, serotonina, acetilcolina e ácido gama-aminobutírico (GABA), que possuem funções efetoras neuroativas. Vários estudos demonstraram que os neurotransmissores derivados do intestino são importantes na regulação do humor e nas doenças psiquiátricas . Além disso, hormônios derivados do intestino produzidos por células enteroendócrinas podem se ligar a receptores nas fibras aferentes vagais. Além disso, os SCFAs também podem atuar diretamente nas fibras vagais aferentes. É possível que a estimulação do nervo vago possa afetar a atividade imunológica no cérebro. Foi relatado que a estimulação do nervo vago diminui a reação neuroinflamatória à administração sistêmica de lipopolissacarídeos e induz o fenótipo microglial antiinflamatório em um modelo de camundongo com DA (BONAZ, 2018; KACZMARCZYK, 2017; MENESES, 2016) .
Além disso, também foi demonstrado que a estimulação do nervo vago libera acetilcolina, que tem um efeito antiinflamatório na expressão gênica de macrófagos na periferia. É possível que a sinalização derivada do intestino através do nervo vago possa estar envolvida na progressão da DA; no entanto, isso ainda não foi testado. Se a sinalização do nervo vago for importante para o envolvimento do GMB na DA, pode ser através de neurotransmissores derivados do intestino, metabólitos ou estimulação vagal mediada por hormônios, alterando as respostas imunes na periferia e no cérebro e subsequente reação alterada das células imunes à patologia amiloide e tau (WANG, 2003).
Os psicobióticos correspondem aos organismos vivos dentro da classe dos probióticos que promovem benefícios para o indivíduo manter a saúde mental em equilíbrio. Além de terem interações com as bactérias comensais, esses organismos têm função de metabolizar substâncias ingeridas e fabricar compostos neuroativos como serotonina, glutamato, BDNF, GABA, entre outros; esses irão agir no eixo intestino-cérebro, exercendo efeito em nível de ansiedade, funções cognitivas, processo de aprendizagem, memória e humor (DE CARVALHO, 2018).
Na relação entre as mudanças alimentares e a DA, os estudos recentes relatam que a utilização abusiva de glúten, açúcar e a baixa ingestão de ácidos graxos poliinsaturados acabam promovendo uma inflamação sistêmica, atingindo assim, o SNC (CRYAN, 2019; HU, 2019).
A perda sináptica e dendrítica e as alterações da plasticidade neuronal se correlacionam melhor com o declínio cognitivo do que a perda neuronal. Estudos sugerem que os oligômeros possam causar problemas cognitivos por interromperem a função sináptica na ausência de neurodegeneração significativa no seu entorno ao alterar as redes neuronais corticais funcionais, levando a uma sub-regulação dessas redes e comprometendo anatomicamente e funcionalmente as áreas cerebrais interconectadas (CHEN et al., 2019; WEBERS 2020).
As conexões entre muitas doenças neurológicas e a composição da microbiota intestinal levou a um interesse maior no uso de probióticos. Um crescente corpo de evidências apoia a ideia que certos probióticos podem impactar positivamente a patogênese de distúrbios neurológicos (KIM et al, 2018).
As bactérias probióticas não apenas modulam as respostas imunes do hospedeiro, mas também criam um ambiente intestinal saudável através do balanceamento da microflora intestinal. A ingestão de probióticos pode restaurar a composição da microflora intestinal para um estado mais favorável de microrganismos benéficos (CHOI et al., 2015).
Alterações internas e externas, particularmente aquelas causadas pelo uso prolongado de antibióticos de amplo espectro, inibem o crescimento de bactérias intestinais sensíveis, permitindo a multiplicação de bactérias patogênicas resultando em disbiose. Nesta situação, o equilíbrio fisiológico normal das bactérias é perturbado, permitindo a acumulação de metabólitos tóxicos (ALSEGIANI 2022; CHIDAMBARAM et al., 2022).
Um estudo clínico confirmou que os sintomas gastrointestinais aparecem frequentemente nos primeiros anos de desenvolvimento de doenças neurodegenerativas (KATSUNO et al., 2018). Patógenos e metabólitos tóxicos entram na circulação sistêmica, levando à desregulação da glicocerebrosidase e à neuroinflamação crônica por meio da desregulação da ativação imunológica, desencadeando a liberação de proteínas neurotóxicas e mal dobradas na circulação sistêmica (KAUR et al., 2021).
Estudos afirmam que há uma comunicação harmoniosa entre o trato gastrointestinal, a microbiota e o sistema nervoso central, possuindo assim, uma relação na qual todos exercem funções e sofrem influência dos mesmos. Assim, quando há um desequilíbrio em algum dos eixos, há consequências que podem levar ao desenvolvimento de doenças (SILVESTRE, 2016).
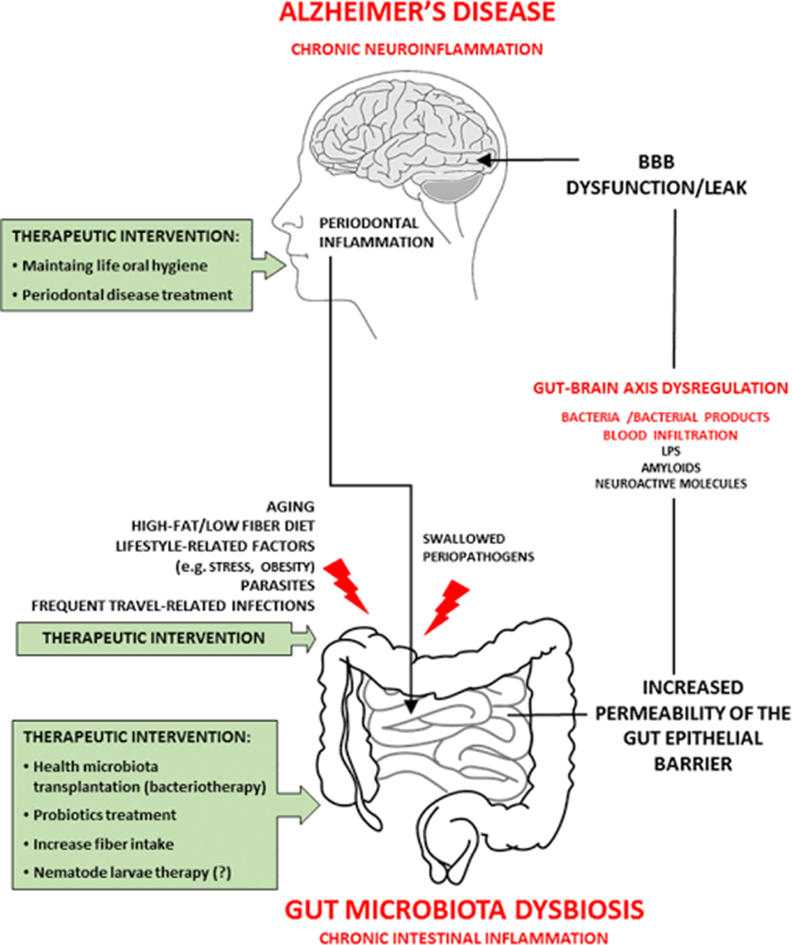
Fonte: Sochocka, 2019.
A perda sináptica e dendrítica e as alterações da plasticidade neuronal se correlacionam melhor com o declínio cognitivo do que com a perda neuronal. Estudos sugerem que os Lipopolissacarídeos (LPS) possam causar problemas cognitivos por interromperem a função sináptica na ausência de neurodegeneração significativa no seu entorno ao alterar as redes neuronais corticais funcionais, levando a uma sub-regulação dessas redes e comprometendo anatomicamente e funcionalmente as áreas cerebrais interconectadas (CHEN et al., 2019; WEBERS 2020).
O SNC pode alterar a composição da microbiota intestinal e a biomassa total através da regulação da saciedade. Logo, as mudanças nos padrões alimentares afetam a capacidade da microbiota intestinal de acessar nutrientes, influenciando a sua composição. Portanto, as bactérias probióticas não apenas modulam as respostas imunes do hospedeiro, mas também criam um ambiente intestinal saudável através do balanceamento da microflora intestinal. Desse modo, a ingestão de probióticos pode restaurar a composição da microflora intestinal para um estado mais favorável para microrganismos benéficos (WANG, 2014).
Agusti (2018) corrobora com os resultados de Wang (2014) e complementa que o SNC também regula a composição da microbiota alterando a permeabilidade epitelial, permitindo assim a penetração de bactérias e facilitando a interação microbiana-hospedeiro na mucosa, isto inclui desregulação do eixo HPA com superprodução de glicocorticóides, alterações nos níveis de metabólitos neuroativos como neurotransmissores, ácidos graxos de cadeia curta e ativação de um ambiente pró-inflamatório que pode causar neuroinflamação.
A inflamação sistêmica pode aumentar a permeabilidade da barreira intestinal e permitir assim a translocação de bactérias comensais, consequentemente gerando mais implicações. Evidências recentes mostraram que bactérias isoladas do intestino tem a capacidade de sintetizar compostos neuroativos incluindo neurotransmissores, muitos dos quais resultam do catabolismo de aminoácidos. Esses compostos incluem GABA, noradrenalina, dopamina e serotonina (PAREDE et al., 2014).
Fatores ambientais como dieta, sono e o desenvolvimento da DA devido à genética contribuem para um ambiente inflamatório na microbiota intestinal, o que leva a mudanças na composição e diversidade ao longo do tempo. Mudanças na microbiota influenciam os metabólitos derivados do microbioma intestinal e a imunidade periférica, alterando a expressão gênica das células imunes periféricas e a liberação de citocinas. Alterações na imunidade periférica, possivelmente metabólitos derivados diretamente do intestino e hormônios derivados do intestino traficados pelo nervo vago podem então alterar o fenótipo da barreira hematoencefálica e os tipos de células do sistema nervoso central (micróglia, astrócitos, neurônios), que podem então modular a amiloidose, tauopatia e neurodegeneração (CHANDRA, 2023).
SONO E MICROBIOTA
Os distúrbios do sono e a disfunção do ritmo circadiano estão fortemente implicados na DA. Os pacientes com DA frequentemente apresentam perturbações no ciclo sono-vigília e ficam cada vez mais acordados à noite e sonolentos durante o dia. Além disso, passam menos tempo no sono de ondas lentas e de movimento rápido dos olhos (REM), ambos críticos para a consolidação da memória e cognição. Como resultado, o sono deficiente e a fragmentação do sono podem prever a DA e a subsequente demência. Além disso, adultos cognitivamente normais que relatam problemas de sono têm maior probabilidade de ter patologia amilóide em seus cérebros na PET scan.
Acredita-se que as oscilações diurnas de Aβ sejam resultado de diferenças na atividade neuronal entre o sono e a vigília. Durante o sono, a atividade neuronal é reduzida e durante a vigília, aumenta. A menor atividade neuronal durante o sono provavelmente leva a uma menor produção de Aβ. A privação do sono agrava a patologia da DA, o que conecta claramente a má qualidade do sono com a progressão da DA. Além disso, a disfunção do ritmo circadiano está implicada na DA. Modelos de DA em ratos mostram disfunção circadiana ao longo do tempo (HOYT, 2022).
Tranah et al. (2011) descobriram que a disfunção circadiana poderia prever o desenvolvimento futuro da DA. Além disso, polimorfismos de nucleotídeo único no gene Clock estão associados a DA. Existem várias linhas de evidência que sugerem que o GMB pode influenciar a qualidade do sono e que a qualidade do sono pode influenciar a composição do GMB. A perturbação do GMB mediada por antibióticos pode resultar em sono NREM fragmentado. Por outro lado, a interrupção do sono pode levar a alterações na composição do GMB.
Voigt et al. (2016) descobriram que camundongos Clock mutantes que interromperam o sono tiveram uma composição de GMB significativamente alterada e menor diversidade taxonômica em comparação aos controles. Este efeito foi exacerbado pelo consumo de álcool, sugerindo que o sono deficiente combinado com outros fatores pode alterar o GMB ainda mais em comparação com o sono deficiente isoladamente. Isto pode sugerir uma hipótese de dois acertos, segundo a qual o sono deficiente pode ter um grande efeito no GMB e predispor a várias condições patológicas.
Poroyko et al. (2016) observaram uma mudança na composição do GMB após a fragmentação do sono ter sido induzida em camundongos do tipo selvagem. Como resultado, estes ratos tiveram inflamação do tecido adiposo e diminuição da sensibilidade à insulina. Além disso, a colonização de camundongos fragmentados pelo sono em animais livres de germes causou esses mesmos fenótipos, implicando que o GMB estava mediando esses efeitos. Da mesma forma, a privação crônica de sono em ratos com 7 dias de idade resultou em alterações na composição do GMB. O mecanismo pelo qual o GMB pode influenciar o sono pode ser através de metabólitos microbianos.
Foi demonstrado que a administração de butirato promove o sono NREM em ratos e camundongos. Além disso, uma percentagem mais elevada de propionato em relação à composição total de SCFA foi associada a um sono infantil humano ininterrupto mais longo. O sono também pode influenciar a inflamação. Geralmente, a perda de sono aumenta as respostas inflamatórias. A conexão entre sono e inflamação pode ser mediada pelo GMB (ZHANG, 2021).
Além da conexão do sono fisiológico interrompido estar associada às alterações do GMB, as condições patológicas do sono também estão associadas às alterações do GMB. A hipóxia intermitente associada à apneia obstrutiva do sono (AOS) está associada à composição do GMB e às alterações na diversidade. Além disso, os pacientes com AOS alteraram a composição do GMB em comparação com os HCs. Da mesma forma, pacientes com insônia e narcolépticos alteraram a composição do GMB em comparação com os HCs.
Por último, várias formulações probióticas/prebióticas melhoram o sono, o que implica que o aumento de bactérias benéficas no GMB pode influenciar os resultados do sono. A evidência sugere que provavelmente existe uma conexão entre o GMB, o sono e a DA. Embora as conexões entre o GMB e o sono e o sono e a DA tenham sido estudadas, a conexão entre o GMB, o sono e a DA em conjunto não foi extensivamente estudada. Esse relacionamento é provavelmente complexo e bidirecional. O sono interrompido pode levar à disbiose intestinal, que pode modular a patologia da DA. Também é possível que a disbiose intestinal possa levar a perturbações do sono, o que pode então modular a patologia da DA. Existe provavelmente um efeito sinérgico destes dois cenários, que contribuem para a patogénese da DA. É imperativo que esta conexão seja estudada para melhor compreender os mecanismos de progressão da DA e para o direcionamento terapêutico da conexão sono-GMB para a DA (LI, 2021; THOMPSON, 2020).
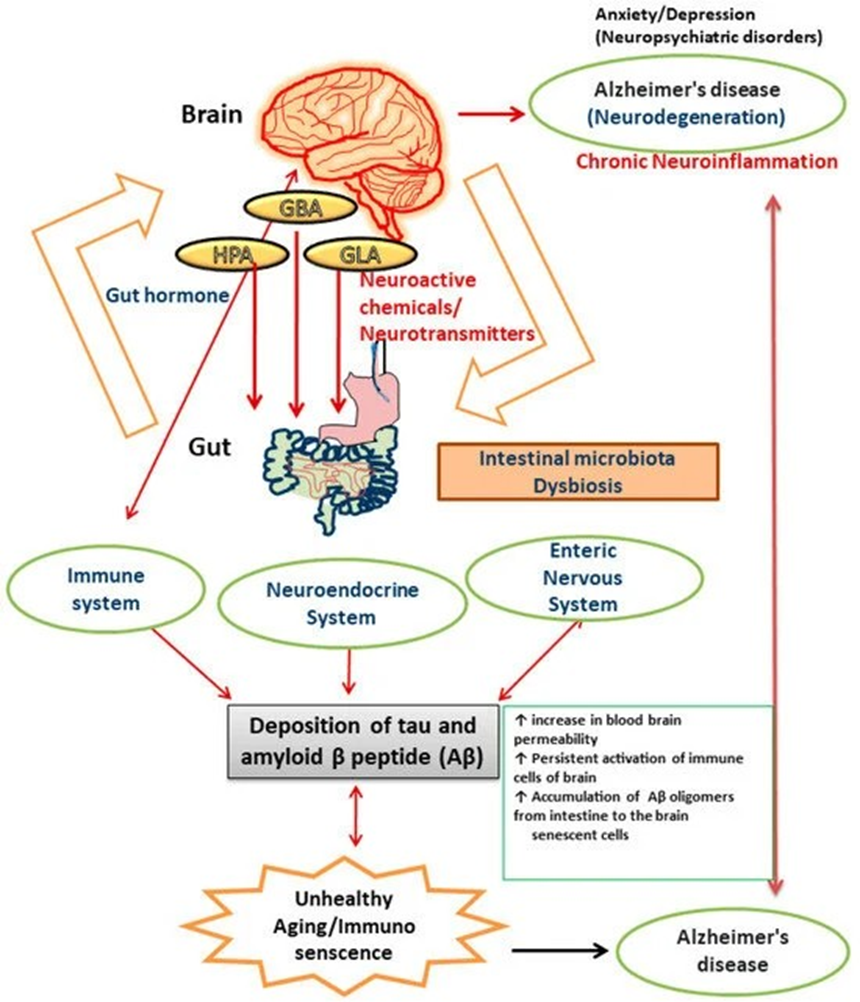
Fonte: Mishra, 2023.
NEUROTRANSMISSORES
Os mecanismos pelos quais as fibras alimentares e o butirato (ácido graxo poliinsaturado) afetam o cérebro podendo envolver fatores neurotróficos como fator de crescimento nervoso (NGF), fator neurotrófico derivado do cérebro (BDNF) e células da glia. Essas pequenas proteínas regulam o crescimento, a sobrevivência e a diferenciação dos neurônios e sinapses no sistema nervoso central e sistema nervoso periférico, desempenhando papéis importantes na aprendizagem, memória e de distúrbios cerebrais. Em estudos experimentais, os efeitos do butirato em fatores neurotróficos potencializam o aprendizado e a memória (SUN et al., 2016).
A produção microbiana de neurotransmissores representa um mecanismo potencial para influenciar diretamente o cérebro e o comportamento. A rota é limitada porque a maioria dos neurotransmissores, incluindo serotonina, dopamina e GABA, normalmente não podem violar a barreira hematoencefálica protetora (ABBOTT, 2006). Modos de ação alternativos incluem a possibilidade de que neurotransmissores derivados de microrganismos cheguem ao cérebro através do nervo vago e de seus neurônios aferentes (FORSYTHE, 2013).
Os precursores de neurotransmissores atravessam a barreira hematoencefálica e depois são convertidos em neurotransmissores ativos, como exemplo as bactérias intestinais que podem influenciar o metabolismo e a disponibilidade do triptofano precursor da serotonina. Isso pode afetar a sinalização serotoninérgica no sistema nervoso central, pois a concentração de triptofano no plasma sanguíneo demonstrou correlação com os níveis cerebrais de serotonina (O`MAHONY et al., 2015). Os pesquisadores apontam que a microbiota intestinal pode produzir ou estimular a produção de neurotransmissores e compostos neuroativos, como serotonina, GABA e dopamina, e que esses compostos podem modular o crescimento bacteriano na microbiota intestinal.
As evidências atuais sugerem uma relação direta da microbiota intestinal à produção de neurotransmissores, o metabolismo e a DA. Estudos em pacientes com DA apresentam diminuição nos níveis de GABA no córtex frontal, temporal e parietal. Nesse contexto, as bactérias que habitam o intestino, como Lactobacillus e Bifidobacterium metabolizam o glutamato, formando o GABA, sendo esse essencial para a cognição do ser humano. Importante ressaltar que 95% da produção de serotonina ocorre no intestino. Dessa forma, foi sugerido que quando o microbioma intestinal encontra-se alterado com redução na concentração dos Lactobacillus e Bifidobacterium, ocorre uma diminuição da produção desse neurotransmissor e, consequentemente, ocorrem alterações cognitivas (HU, 2019).
O papel fundamental da serotonina no eixo intestino-cérebro tem sido extensivamente revisto, indicando que perifericamente esta envolvido na modulação do sistema imune do intestino, secreções gastrointestinais, motilidade, sensibilidade visceral e centralmente no humor e cognição (O’MAHONY et al., 2015).
A serotonina está envolvida no comportamento, aprendizagem, apetite e homeostase da glicose produzida no cérebro humano e nas células intestinais enteroendócrinas. Através do eixo cérebro-intestino, é possível verificar que a microbiota tem influência sobre o funcionamento do sistema nervoso, da mesma forma que o sistema nervoso tem atuação sobre o intestino, estabelecendo uma relação moduladora. Com base nisso, pode-se afirmar que, alteração na composição e/ou funcionamento da microbiota, podem induzir o aparecimento de doenças, inclusive as de teor neurodegenerativo mostrando a relação direta entre o papel da microbiota intestinal e a DA (CRYAN, 2019).
Apesar dos avanços ainda são necessários estudos prospectivos para melhorar a partir de uma fase descritiva da pesquisa, para melhor elucidar e descomplicar a avaliação da disbiose e, fora isso, concluir sobre os tipos de disbiose (por exemplo, perda de bactérias benéficas, crescimento excessivo de bactérias patogênicas, redução de bactérias diversidade) e se cada tipo tem pesos iguais ou distintos para desencadear ou agravar diferentes categorias de doenças (BRÜSSOW, 2020 apud NAUFEL, 2023).
Fonte: Naufel, 2023.
Vias biossintéticas e metabólicas da serotonina e quinurenina a partir do triptofano. O aminoácido essencial triptofano forma a base para a síntese da serotonina (5-HT). Sua via metabólica dedutiva consiste na via da quinurenina. Todos os circuitos serotoninérgicos do cérebro começam nos núcleos da rafe, uma coleção de neurônios produtores de serotonina, localizados no tronco cerebral, na altura da ponte, e têm eferentes que se conectam com todo o neocórtex, sistema límbico (do qual amígdala e hipocampo), diencéfalo, cerebelo e sistema nervoso periférico/autônomo.
Evidências crescentes sugerem que o microbioma intestinal (GM) desempenha um papel fundamental na patogênese da DA através do eixo microbiota-intestino-cérebro (MGB). Alterações na composição e diversidade dos GM foram observadas tanto em modelos animais quanto em pacientes humanos com DA. A disbiose GM tem sido implicada no aumento da permeabilidade intestinal, no comprometimento da barreira hematoencefálica (BHE), na neuroinflamação e no desenvolvimento de características da DA.
Uma maior elucidação do papel do GM na DA poderia abrir caminho para o desenvolvimento de métodos preditivos holísticos para determinar o risco de DA e a progressão da doença. Além disso, o acúmulo de evidências sugere que a modulação GM poderia aliviar os sintomas adversos da DA ou servir como medida preventiva. Além disso, evidências crescentes mostram que o Diabetes Mellitus Tipo 2 (DM2) é frequentemente comórbido com a DA, com alterações comuns do GM e resposta inflamatória, o que poderia traçar o desenvolvimento de intervenções de tratamento relacionadas ao GM para ambas as doenças. Concluímos explorando o potencial terapêutico do GM no alívio dos sintomas da DA e na redução do risco (KRISHAA, 2023).
O mecanismo de ação de um ISRS é bloquear o SERT, evitando assim a recaptação da serotonina após sua liberação da fenda sináptica de volta ao neurônio pré-sináptico. A via da quinurenina provoca a formação de metabólitos neurotóxicos e neuroprotetores. O ácido cinurênico é formado principalmente nos astrócitos e é um antagonista eficaz do receptor NMDA, prevenindo a liberação intracelular abundante de cálcio e, consequentemente, a excitoxicidade. Por outro lado, a 3-hidroxiquinurenina é conhecida como um potente estressor oxidativo e doador de radicais livres, levando a danos mitocondriais e a criação de ERO. Uma função neurotóxica semelhante foi atribuída ao ácido quinolínico, um agonista do receptor NMDA, com função oposta em comparação ao ácido cinurênico. É relevante ressaltar que o ácido quinolínico é formado principalmente na micróglia (AALDIJK, 2022).
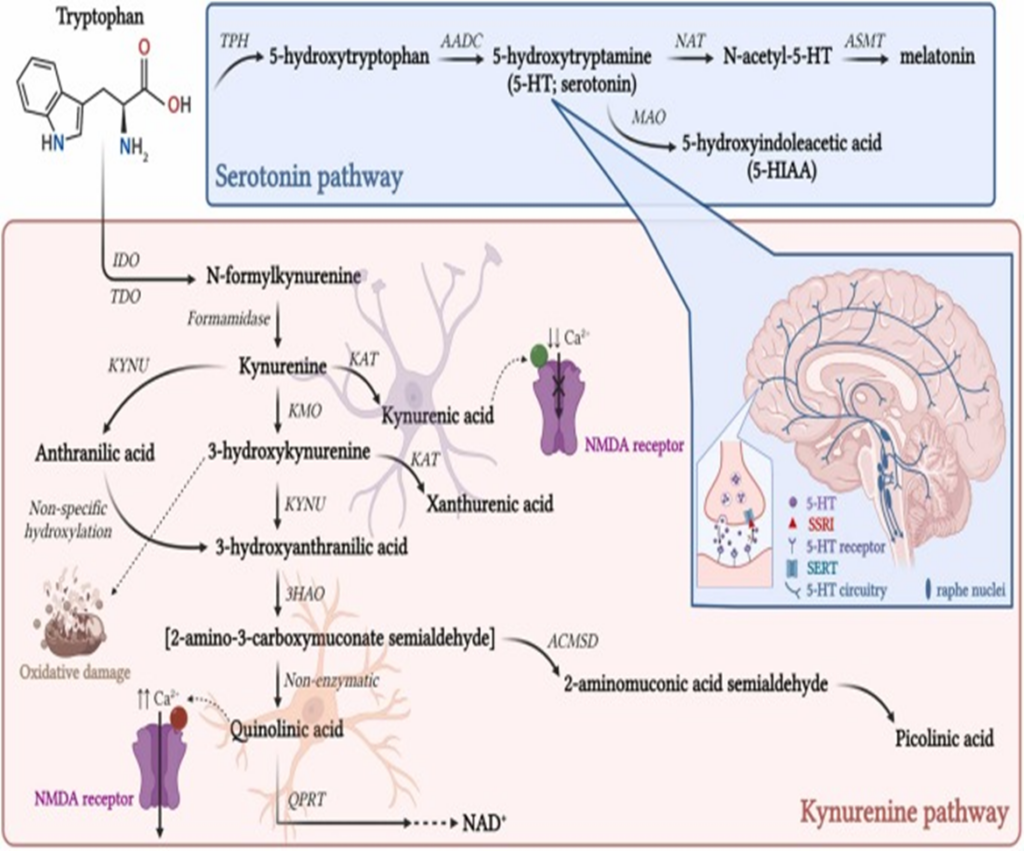
Fonte: Aaldijk, 2022.
A disbiose intestinal contribui para a patogênese da DA e as cepas de Bacteroides são seletivamente elevadas na microbiota intestinal da DA. No entanto, ainda não se sabe quais espécies de Bacteroides e como seus metabólitos desencadeiam as patologias da DA. Bacteroides fragilis e seus metabólitos ácido 12-hidroxi-heptadecatrienóico (12-HHTrE) e Prostaglandina E2 (PGE2) ativam a microglia e induzem a patogênese da DA em camundongos transgênicos neuronais C / EBPβ.
A recolonização de camundongos transgênicos Thy1-C/EBPβ pré-tratados com coquetel de antibióticos com amostras fecais de pacientes com DA provoca patologias de DA, associadas à regulação positiva da via C/EBPβ/Asparaginil endopeptidase (AEP), ativação da microglia e distúrbios cognitivos em comparação com camundongos que recebem fezes de doadores saudáveis transplante de microbiota (FMT).
A análise de sequenciamento de 16S rRNA microbiano mostra maior abundância de Bacteroides fragilis pró-inflamatórios em camundongos AD-FMT. A caracterização dos componentes ativos dos soros e cérebros dos camundongos transplantados revelou que tanto o 12-HHTrE quanto a PGE2 ativam a microglia primária, ajustando-se ao enriquecimento de metabólitos de ácidos graxos poli-insaturados (PUFA) identificados pela metabolômica. Surpreendentemente, a recolonização com Bacteroides fragilis vivos, mas não mortos, provocou patologias de DA em camundongos transgênicos Thy1-C / EBPβ, assim como o tratamento com 12-HHTrE ou PGE2 sozinho (XIA, 2023).
Os estressores psicológicos e físicos crônicos interrompem as adaptações naturais do corpo ao estresse resultando em um “desgaste” geral no corpo, levando à hipercortisolemia, à regulação do eixo hipotálamo-hipófise-adrenal (HPA), à elevação das citocinas proinflamatórias e quimiocinas, à plasticidade sináptica reduzida, à micróglia ativada persistentemente e à microbiota intestinal disbiótica (WESTFALL et al., 2019).
Evidências indicam que o estresse oxidativo pode provocar danos na atividade das enzimas envolvidas no metabolismo da glicose, levando à disfunção sináptica e eventual morte neuronal. Além disso, o estresse oxidativo e a resistência à insulina podem levar a disfunção mitocondrial prejudicando a produção de energia, visto que duas das etapas para a produção de ATP ocorrem na mitocôndria (BUTTERFIELD, 2019).
Segundo Lee (2022), as deficiências precoces da atividade lisossômica, disfunção autofágica e acúmulo da proteína Aβ ocorre antes da deposição das placas amilóides extracelulares. Além disso, identificou-se substratos de autofagia incompletamente digeridos acumulando-se progressivamente nos neurônios afetados no estágio inicial da DA através das técnicas de imagem e histoquímica, levando assim a uma reconsideração da sequência de eventos convencionalmente aceita na formação das placas Aβ na DA. Para o autor, considerar a DA como uma doença autoimune levanta a possibilidade de identificar fatores que modulam as principais citocinas associadas à imunidade funcionando como moléculas endógenas para direcionar outras vias de regulação imunológica no cérebro.
Quando as bactérias estão presentes no cérebro, a proteína Aβ é um dos principais contribuintes para a resposta imune. Devido ao ataque do sistema imunológico do cérebro ao próprio órgão que deveria estar defendendo, surge a hipótese que a DA é uma doença autoimune. No modelo experimental de DA, a proteína Aβ ajuda a proteger e fortalecer o sistema imunológico, mas, também desempenha um papel central no processo autoimune (MEIER‐STEPHENSON, 2022). Acredita-se, portanto, que a proteína Aβ não é uma proteína produzida de forma anormal, mas sim uma molécula de ocorrência normal que faz parte do sistema imunológico do cérebro (FRIZONE, 2022).
Lee e colaboradores (2022), identificaram uma possível disfunção dos lisossomos, organelas celulares que compõem os neurônios e têm como função “digerir” a proteína Aβ acumulada. As descobertas recentes acrescentam evidências de que a acidificação lisossomal e a desregulação do complexo mitocondrial são alvos comuns de disfunções imunometabólicas.
Durante o envelhecimento, as mitocôndrias e os lisossomos liberam cada vez mais ERO. O aumento nos níveis de ERO também decorre da ativação microglial relacionada ao Aβ, inflamação e ligação de metais redox (SOLLEIRO-VILLAVICENCIO, 2018).
A inflamação crônica devido a distúrbios metabólicos também pode se propagar para o SNC pela entrada de moléculas inflamatórias na área do tronco cerebral, o que pode afetar os núcleos colinérgicos como o tegmento pontino laterodorsal e núcleo tegmental pedunculopontino que têm eferências para o córtex e colinérgicos do prosencéfalo basal (ZOLKIPLI-CUNNINGHAM, 2017).
Os resultados sugerem que a inflamação periférica e central estão interrelacionadas ao comprometimento cognitivo e à disfunção colinérgica (CHANG, 2019).
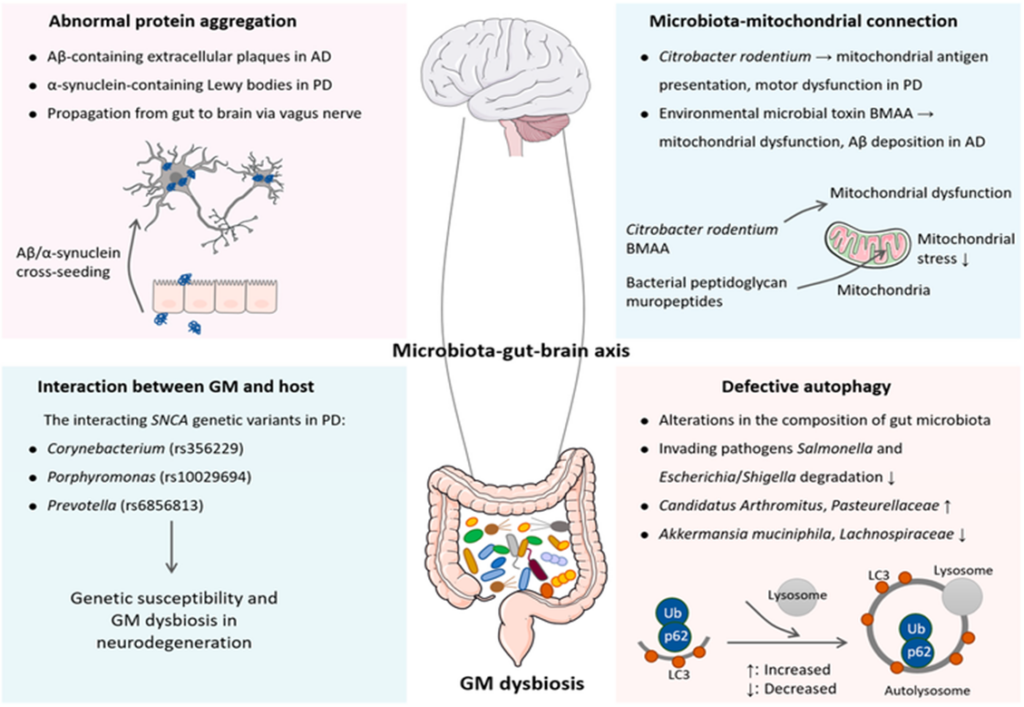
Fonte: Bi et al., 2023.
Mondanelli et al. (2019) concluíram que as vias metabólicas do L-triptofano e da L-arginina são fundamentais para o controle da imunidade inata e que a interação entre essas duas vias pode ser central para a reprogramação da função das células imunológicas em doenças de base imunológica. Além disso, tanto o L-triptofano e L-arginina demonstraram ser inibidores diretos da oligomerização Aβ (além de outros mecanismos de regulação da imunidade inata), sugerindo a capacidade dos aminoácidos de permitir inibidores multissítios da progressão da DA. Estas descobertas demonstram que o metabolismo alterado do L-triptofano e da L-arginina em diversas regiões do cérebro com DA merece uma investigação mais aprofundada para compreender o seu papel na patogênese.
Concomitantemente, os oligômeros Aβ são diretamente imunotóxicos interagindo com a micróglia e os astrócitos mediada em parte por MCP-1, ICAM-1), distorcendo a ativação microglial em direção ao fenótipo M1 pró-inflamatório, promovendo a liberação de citocinas pró-inflamatórias (IL-1β, IL-6, TNFα) e suprimindo a liberação de citocinas anti-inflamatórias (IL-3, IL-4, IL-10, IL-13). Por sua vez, isso promove agregação neurotóxica de Tau (com instabilidade dos microtúbulos) e dano mitocondrial, levando ao aumento de ERRO membranotóxicas (peróxidos, superóxido, radical hidroxila, oxigênio singlet). Finalmente, isto culmina na morte celular por necrose e apoptose, com expressão de sintomas associados na forma de cognição e processamento desordenado de informações na memória de curto prazo (MEIER-STEPHENSON, 2022).
Ligantes do receptor acoplado à proteína G (GPCR), que preferencialmente ativam as vias de sinalização da proteína G ou da β-arrestina, estão levando ao desenvolvimento de medicamentos com eficácia superior e efeitos colaterais reduzidos em doenças cardíacas, controle da dor e distúrbios neuropsiquiátricos. Embora os GPCRs estejam implicados na fisiopatologia da DA, a sinalização distorcida do GPCR é uma área de investigação amplamente inexplorada na DA (HUANG, 2022).
Em estudo anterior, Huang (2022), demonstrou que a sinalização de β-arrestina mediada por GPR3 modula a geração de Aβ in vitro e que a deficiência de Gpr3 melhora a patologia de Aβ in vivo. No entanto, camundongos deficientes em Gpr3 apresentam vários fenótipos adversos, incluindo comportamento semelhante à ansiedade elevada, fertilidade reduzida e comprometimento da memória, que estão potencialmente associados à sinalização prejudicada da proteína G. As alterações na patologia amilóide são acompanhadas por robusta hipertrofia microglial e astrocítica, o que sugere uma resposta glial protetora que pode limitar o desenvolvimento da placa amilóide em camundongos GPR3 com tendência à proteína G.
Em modelos animais de tauopatia ocorre o desenvolvimento da resposta imune inata e adaptativa no cérebro, caracterizada por um aumento acentuado no número de células T, especialmente linfócitos T citotóxicos, e correlacionada com a extensão da perda neuronal. A depleção de células T em camundongos Tau (P301S) via administração intraperitoneal de anticorpos anti-CD4 e anti-CD8 é suficiente para melhorar a atrofia cerebral e o comportamento, sugerindo que a infiltração de células T no parênquima cerebral é um passo fundamental na neurodegeneração. A infiltração de células T CD8+ em neurônios derivados de células-tronco, astrócitos e culturas de micróglia também leva ao aumento da neurodegeneração. No entanto, ainda não está claro se a infiltração de células T se correlaciona com a progressão da patologia da DA. Neste trabalho, dados de sequenciamento de RNA de núcleotídeo único (snRNA-seq) obtidos do cérebro de pacientes com DA e controles pareados foram explorados com o objetivo de caracterizar as respostas imunes inatas e adaptativas em diferentes estágios patológicos (COSTA, 2024).
A autoimunidade é considerada um distúrbio crônico da imunidade adaptativa, e não da imunidade inata, pois a imunidade adaptativa é altamente específica, duradoura e “lembrada” por meio de respostas de anticorpos, enquanto a resposta imune inata é imediata, inespecífica e não lembrada.
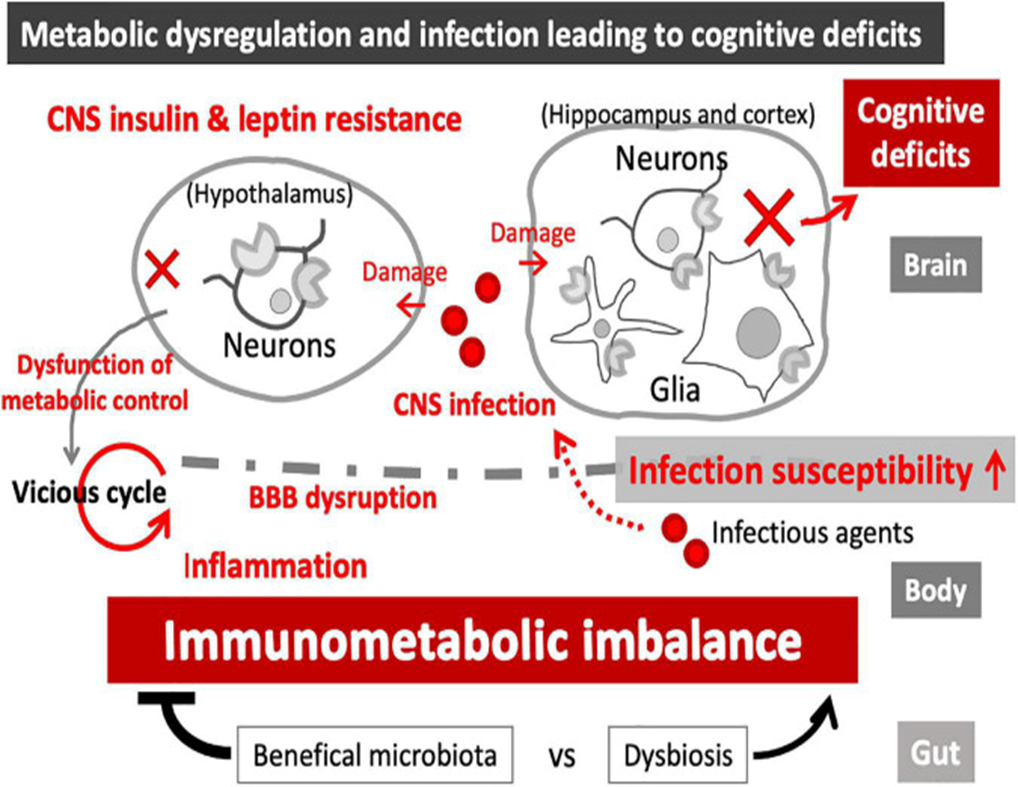
Fonte: Shinjyo, 2021.
A crescente evidência sobre a existência de uma correlação estreita e bidirecional entre a microbiota intestinal e o cérebro indica que o desequilíbrio intestinal pode desempenhar um papel vital no desenvolvimento, função e distúrbios do sistema nervoso central. Os mecanismos de interação entre a microbiota intestinal na atividade neuronal são amplamente discutidos. Várias vias potenciais estão envolvidas com o eixo cérebro-intestino-microbiota, incluindo o nervo vago, vias endócrinas, imunológicas e bioquímicas. A disbiose intestinal tem sido associada a distúrbios neurológicos de diferentes maneiras que envolvem ativação do eixo hipotálamo-hipófise-adrenal, desequilíbrio na liberação de neurotransmissores, inflamação sistêmica e aumento da permeabilidade da barreira intestinal e hematoencefálica (NAUFEL, 2023).
ENDOFENÓTIPO IMUNOMETABÓLICO
Os metabólitos imunológicos específicos do sexo e vias genéticas estão associados à patogênese e progressão da DA. Indivíduos do sexo feminino com DA exibem endofenótipos de imunometabolismo microglial caracterizados por diminuição do metabolismo do glutamato e atividade elevada da via da interleucina-10. Indivíduos do sexo feminino com DA mostraram uma mudança nas comunicações célula-célula mediadas por glutamato entre neurônios excitatórios para micróglia e astrócito.
Os resultados apontam para uma nova compreensão da base molecular da predominância feminina na DA, e garantem futuras validações independentes com coortes de pacientes etnicamente diversas para estabelecer uma provável relação causal do imunometabolismo microglial nas diferenças sexuais na DA. Na conclusão dos autores, as diferenças na regulação do imunometabolismo parecem desempenhar um papel importante nas manifestações biológicas específicas do sexo na DA, em particular na forma específica da micróglia. Esta nova compreensão poderia ajudar a melhorar o desenvolvimento de terapêuticas, ensaios clínicos e tratamento de pacientes com DA (O’MAHONY, 2024).
No estudo posterior de O’mahony (2025), uma proteína chave que pode desempenhar um papel durante a menopausa e pode contribuir para a elevada incidência da DA em mulheres é a Forkhead box O3 (FOXO3). Alterações na expressão de FOXO3 foram observadas nas pacientes com DA (PRADHAN et al., 2020), e está intimamente ligada ao estradiol na regulação da longevidade e do metabolismo celular (SANESE et al., 2019). Esta conexão sugere que o FOXO3 poderia servir como uma ligação metabólica entre a DA e a menopausa. Este estudo revisou o papel potencial do FOXO3 na progressão da DA, destacando suas interações com vias hormonais e metabólicas. Compreender as causas subjacentes das alterações específicas do sexo na DA e desenvolver tratamentos personalizados para ambos os sexos poderia melhorar significativamente a qualidade de vida dos pacientes acometidos.
O envelhecimento também está associado a um declínio significativo na expressão de receptores de estrogênio, particularmente ERβ, que é marcadamente reduzido no hipocampo e nas mitocôndrias neuronais nos cérebros femininos afetados pela DA. Este declínio pode estar correlacionado com a diminuição da eficiência mitocondrial, comprometimento do metabolismo da glicose, aumento do dano oxidativo e redução da resiliência metabólica neuronal. Acredita-se que tais perturbações metabólicas contribuam para o aumento da vulnerabilidade das mulheres na pós-menopausa a DA. A compreensão do papel do estradiol e dos seus receptores no metabolismo cerebral fornece informações valiosas sobre as suas funções neuroprotetoras, com o objetivo principal de desenvolver intervenções direcionadas para modular a disfunção metabólica e reduzir o risco de DA em mulheres idosas (RASIC-MARKOVIC et al., 2024; KLINGE, 2020).
À medida que o estradiol diminui, ocorre uma redução correspondente na captação de glicose, conforme evidenciado por estudos de tomografia por emissão de pósitrons com fluordesoxiglicose (FDG-PET). Estes estudos revelam hipometabolismo em regiões do cérebro, como o hipocampo e o córtex cingular posterior, que são particularmente vulneráveis a patologia da DA. Além disso, a disfunção mitocondrial ligada a deficiência de estradiol agrava o estresse oxidativo e prejudica a produção de ATP, o que acaba por afetar a atividade sináptica e a resiliência neuronal. Portanto, não há dúvida de que a disfunção mitocondrial é uma marca registrada na patologia da DA (GORHAM et al., 2023; SOUSA et al., 2023; BARRETO, 2023).
A análise histopatológica do tecido cerebral post-mortem em mulheres na pós-menopausa não mostrou alterações nos níveis de andrógenos e estrogênios. Em contraste, as mulheres com DA tinham níveis baixos de andrógenos e estrogênios, independentemente da idade das pacientes. No cérebro masculino, o envelhecimento foi correlacionado com baixos níveis de andrógenos e estrogênios (GILLETT, 2023; ROSARIO, 2021).
No estudo de Ocañas (2023), houve diferenças sexuais identificadas na micróglia jovem do hipocampo, com a maioria dos genes diferencialmente expressos (DEGs) restritos aos cromossomos sexuais. As diferenças sexuais codificadas cromossomicamente surgiram com o envelhecimento. Os DEGs sexuais identificados na velhice eram principalmente tendenciosos para o sexo feminino e enriquecidos em assinaturas microgliais senescentes associadas a doenças.
Estes dados sugerem que a micróglia feminina adota fenótipos senescentes associados a doenças no envelhecimento do hipocampo mesmo na ausência da doença e em maior extensão do que os homens. Este fenótipo microglial sexualmente divergente pode explicar a diferença na suscetibilidade e na progressão da DA. Estudos futuros precisarão explorar as diferenças sexuais na heterogeneidade microglial em resposta a patologia da DA e determinar como os reguladores específicos do sexo (cromossomos e hormônios) provocam esses efeitos.
Segundo Ocañas (2023), a microglia adota estados funcionais heterogêneos definidos por sua motilidade, capacidade fagocítica, programação transcricional, expressão de marcadores de superfície celular e fenótipo secretor. Em última análise, a proliferação microglial pode levar à senescência replicativa, que possui um fenótipo que reflete o estado associado à doença observado em modelos de camundongos com DA. As características das células senescentes incluem: (1) parada do ciclo celular, (2) resistência à apoptose e (3) regulação positiva dos fatores do fenótipo secretor associado à senescência (SASP). Nossa hipótese é que a microglia feminina tem uma carga maior de inflamação e senescência em comparação com os homens, contribuindo assim para os preconceitos sexuais observados nas doenças cerebrais relacionadas à idade.
A reatividade microglial sexualmente divergente pode contribuir para a prevalência e progressão feminina de doenças cerebrais relacionadas à idade, como a DA. Para fornecer uma ferramenta para avaliar as diferenças sexuais microgliais no campo, os níveis normalizados de expressão gênica para as análises transcriptômicas e translatômicas foram implantados em uma interface da Web para pesquisa.
Cabe ressaltar que a micróglia conforme descrito no capítulo 4, são células efetoras da imunidade inata no cérebro e altamente plásticas desempenhando uma ampla gama de funções especializadas incluindo a fagocitose e a remoção de agregados amilóide e Tau que impulsionam a neurodegeneração. Essas funções imunológicas requerem alta demanda energética, que é regulada pelas mitocôndrias.
Refletindo isso, a micróglia demonstrou ser flexível do ponto de vista metabólico, reprogramando sua função mitocondrial após ativação inflamatória para atender às suas demandas energéticas. No entanto, os fatores de risco genético associados a DA prejudicam a programação metabólica microglial. Além disso, foi demonstrado que o descarrilamento metabólico causa disfunção imunológica inata na DA. Estas descobertas sugerem que a imunidade e a função metabólica são processos intrinsecamente ligados, e o direcionamento do metabolismo microglial oferece uma janela de oportunidade para o tratamento terapêutico da DA (FAIRLEY, 2021).
As heterogeneidades endofenotípicas do imunometabolismo também mostraram um padrão específico de sexo na DA a partir de metabólitos, vias e tipos de células. A diminuição da abundância de N-acetilglutamato e N-acetil-aspartil-glutamato (NAAG) foi observada de forma consistente com 10.000 coortes de mulheres com DA.
A micróglia apresentou diminuição no metabolismo do glutamato e da glutamina em mulheres com DA, o que potencialmente influencia as comunicações célula-célula entre a micróglia e o neurônio. As análises de imagens PET com fluoroglutamina mostraram nos gliomas masculinos uma captação de glutamina significativamente maior, e os astrócitos masculinos exibiram maior sensibilidade à inibição da glutaminase 1 (GLS1) (GLENON, 2013; PROPIK, 2014).
Esses achados fornecem evidências preliminares de que o N-acetilglutamato pode oferecer um potencial biomarcador específico do sexo para a DA. A descoberta demonstrou que o hormônio sexual modulado pelo estrogênio induziu neuroinflamação na progressão da DA. Esse estudo indica que o estrogênio medeia a comunicação entre o neurônio e a micróglia em indivíduos com DA (PONNUSAMY, 2022; SARTORI, 2019).
Nas mulheres com DA, foi demonstrado uma redução relacionada ao metabolismo dos hormônios esteróides e as vias imunes inatas, como as vias e a sinalização da IL-6, mas nenhuma alteração foi observada nos homens. A análise revelou que as mulheres também apresentam uma carga mais grave da patologia consistente com estudos anteriores de que exibem mais marcadores neuropatológicos do que os homens, incluindo estágios mais elevados de Braak para emaranhados e escores amilóides (HU et al, 2021).
Os dados foram gerados a partir de amostras humanas e a validação experimental é necessária em estudos futuros de metabólitos específicos do sexo, como os metabólitos do GPC e o metabolismo do glutamato, em coortes humanos sem e com a DA. Por fim, sabe-se que fatores genéticos, incluindo a inativação parcial do cromossomo X e hormônios, têm impacto nas diferenças sexuais na DA (HOU, 2023).
A micróglia expressa genes necessários tanto para o metabolismo glicolítico como oxidativo e pode alternar entre estes programas metabólicos em resposta a estímulos inflamatórios (GHOSH, 2018; ZHANG, 2014). Estas descobertas indicam que os fatores de risco genéticos estão intrinsecamente associados as funções metabólicas mitocondriais na micróglia. No entanto, mais pesquisas são necessárias para elucidar as vias envolvidas na regulação metabólica na micróglia, o que pode, por sua vez, pode auxiliar na identificação de alvos moleculares para modular o comprometimento imunometabólico (HOLLAND, 2018; WANG, 2016).
A micróglia mostrou diferenças entre os sexos no imunometabolismo na DA através de dados de snRNA-seq. Por exemplo, o córtex feminino com a DA apresentou maior proporção de micróglia em comparação aos controles (p = 0,034 e FDR = 0,275), embora não seja altamente significativo. Uma possível explicação para a falta de valores altamente significativos pode ser causada pelas baixas populações microgliais nos conjuntos de dados atualmente disponíveis (DU, 2008).
Micróglia e astrócito em mulheres com DA exibiram atividade de sinalização de IL-10 elevada em comparação com controles. No entanto, os indivíduos do sexo masculino com DA apresentam diminuição da atividade de sinalização da IL-10 em comparação com os controles do sexo masculino. A IL-10 exibe um efeito anti-inflamatório em macrófagos. Esse efeito é mediado pela reprogramação metabólica da glicólise e do ciclo do ácido tricarboxílico, facilitando a mitofagia para remover as mitocôndrias disfuncionais (LUSTBADER, 2004).
Foi demonstrado que o bloqueio da via anti‐inflamatória da IL-10 melhora a atividade da micróglia fagocítica e limita a amiloidose cerebral em modelos de camundongos com DA. Portanto, é crucial considerar diferenças específicas de sexo nas respostas anti‐inflamatórias da IL‐10 ao desenvolver futuras estratégias terapêuticas para a DA (MANCZAK, 2011).
Estudos experimentais sugerem um papel dos esteróides sexuais para influenciar o acúmulo de β-amilóide e também para modular as respostas neuronais à lesão. No neuroblastoma murino e nas culturas neuronais primárias, e nas células hipotalâmicas de ratos, o tratamento com testosterona aumentou a clivagem da proteína precursora β-amilóide para aumentar a secreção de fragmentos não amiloidogênicos. Em experimentos em neurônios do hipocampo de ratos, a testosterona e o estradiol tiveram efeitos diferenciais na clivagem da proteína Tau, e a testosterona foi mais eficaz na prevenção da morte celular induzida por β-amilóide (SONG, 2020; IMTIAZ, 2017; SAVOLAINEN-PELTONEN, 2019; SHUMAKER, 2003; TOLPPANEN, 2018).
Em um modelo de camundongo transgênico superexpressando a proteína precursora de amilóide, a regulação negativa da expressão da aromatase aumentou a testosterona e reduziu as concentrações de estradiol com a formação de placas no cérebro, inferindo um papel da testosterona em vez do estradiol para proteger contra a DA (SHAO, 2012). No entanto, outros estudos relatam um papel dos estrogênios e da progesterona na proteção dos neurônios do hipocampo de ratos cultivados contra a toxicidade do glutamato (GILLIES, 2010). O estradiol (e também a estrona e o estriol) inibiram a formação de β-amilóide in vitro (CUI, 2013).
De acordo com Bianchi (2025), a testosterona pode ter efeitos protetores no cérebro, retardando o desenvolvimento das múltiplas patologias encontradas em homens com DA. Estudos epidemiológicos de homens de meia idade e após os 65 anos, associam concentrações mais baixas de testosterona a maior prevalência e incidência de declínio cognitivo e demência. Estudos observacionais de homens com câncer de próstata associaram a diminuição da função cognitiva e maior risco de demência. No entanto, a evidência de causalidade permanece indefinida. Estudos menores de intervenção com testosterona, utilizando diferentes medidas da função cognitiva, produziram resultados inconsistentes.
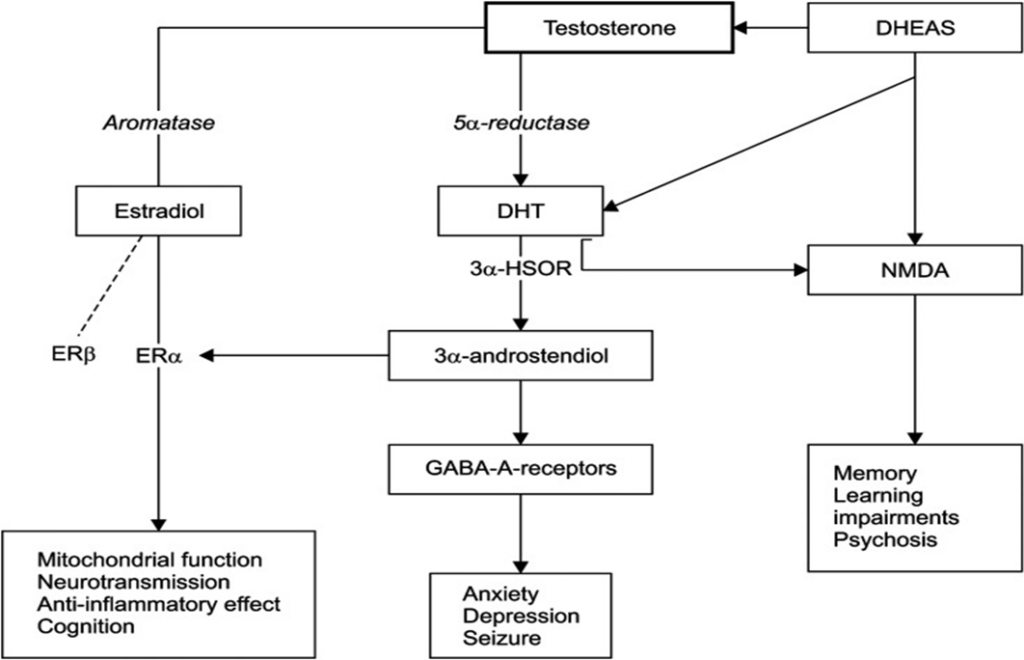
Fonte: Bianchi, 2025.
A testosterona, através do efeito da α-redutase, é reduzida a DHT. O DHT está no Androstenediol, do qual os metabólitos 3α- e 3β-diol têm um efeito fraco, enquanto são mais ativos em Erα e Erβ. O 3α-diol ativa os receptores GABA que regulam a ansiedade, a depressão e as convulsões. A testosterona também é aromatizada em 17β-estradiol que, ativando Erα e β, estimula a função mitocondrial, a neurotransmissão e o efeito antiinflamatório com consequente melhora da cognição ativando os receptores NMDA que regulam a memória, dificuldades de aprendizagem (BIANCHI, 2025).
A alta expressão dos genes codificados no cromossomo X (Kdm6a, Eif2s3x, Xist) em mulheres quando comparadas aos homens (Kdm6a codifica o modificador de histona desmetilase 6A específico da lisina, que desmetila seletivamente o H3K27, desempenhando um papel importante na modulação do epigenoma. Recentemente, foi demonstrado que o Kdm6a desempenha um papel importante na função cognitiva dependente do hipocampo. É lógico que a expressão sexualmente dimórfica de Kdm6a pode estabelecer diferenças na paisagem epigenômica microglial específica do sexo e mediar respostas diferenciais ao envelhecimento e à patologia da doença (SHAW, 2023).
Dentro das vias de doenças e funções biológicas reguladas diferencialmente pelo sexo, identificamos diversas vias envolvidas em processos migratórios e de adesão, sugerindo diferenças sexuais na motilidade da microglia envelhecida. O exame dos reguladores a montante ativados diferencialmente por sexo identificou muitos genes envolvidos na resposta pró-inflamatória (isto é, IFNG, TNF, IL1B) em micróglias femininas antigas. Foi demonstrado que o interferon-gama (IFNG) estimula a microglia e causa uma reação mais responsiva ao estímulo secundário, além de induzir a proliferação microglial e alterar o disparo neuronal do hipocampo (TA, 2019).
Assim, uma maior indução de IFNG em micróglias femininas idosas provavelmente as leva mais rapidamente à senescência e subsequentemente resulta em maior suscetibilidade à patologia da DA (ou seja, amilóide). Fatores de necrose tumoral (TNFs), como o TNFα, são reguladores mestres da liberação de citocinas pró-inflamatórias e demonstraram fornecer feedback positivo autócrino para aumentar a produção de TNFα na microglia. A maior ativação das vias do TNF pode levar a essa divergência sexual da sinalização inflamatória microglial através de ciclos de feedback positivo autócrinos, que podem ser ainda mais exacerbados com o aumento da idade.
Outro importante regulador a montante que é mais induzido na microglia feminina antiga do que na microglia masculina antiga é o SPI1, que codifica o fator de transcrição PU.1, um regulador mestre do desenvolvimento e ativação microglial. O SPI1 também é um fator de risco genético para DA e tem sido associado à proliferação de macrófagos. Como o SPI1 é um fator crítico de transcrição microglial, mesmo pequenas alterações na atividade podem levar a diferentes suscetibilidades a DA (JONES, 2022; PIMENOVA, 2021).
O fator estimulador de colônias 1 (CSF1), outro regulador central da identidade microglial, mostrou maior ativação na microglia feminina em comparação com a microglia masculina antiga. O CSF1 regula a sobrevivência, proliferação, diferenciação e função da microglia e de outros macrófagos. Como o CSF1 induz a proliferação , a sinalização aprimorada do CSF1 pode levar a fenótipos senescentes mais exagerados na microglia feminina e à neuroinflamação concomitante.
A micróglia feminina também mostrou uma maior ativação das vias reguladoras a montante associadas a DA (APP, TREM2), sugerindo um mecanismo potencial para a progressão da doença na DA com tendência feminina. Além disso, dois reguladores a montante do modificador do epigenoma (TET2, DNMT3A) demonstraram ser mais ativados na microglia feminina idosa em comparação com os machos idosos, fornecendo um potencial mecanismo epigenômico pelo qual essas diferenças sexuais na microglia podem ser perpetuadas e mantidas. Além das atividades individuais desses reguladores a montante que são mais altamente ativados na antiga microglia feminina, vários reguladores fornecem feedback positivo dentro de uma rede, de modo que a ativação de um regulador amplifica ainda mais o efeito de outros reguladores (DE, 2021).
DOENÇA AUTOIMUNE
À medida que surgem novos alvos biomoleculares para DA, há uma tendência para considerá-los mutuamente exclusivos e em competição, culminando em declarações de que, uma vez que a “hipótese amilóide está morta”, necessita de ser substituída por teorias completamente diferentes. A incorporação de mecanismos de agregação de proteínas em modelo imunopático de DA mais abrangente poderia atingir esse objetivo – um objetivo que poderia ser alcançado reposicionando o papel de Aβ como um imunopeptídeo (WEIVER, 2021).
Em estudo posterior, Weiver (2023) nomeou a DA como doença autoimune (AD2) em resposta aos eventos estimuladores de padrões moleculares associados a patógenos/danos desencadeando uma cascata de imunidade inata na qual a Aβ exibe ação imunomoduladora/antimicrobiana. No entanto, as propriedades antimicrobianas de Aβ resultam num ataque mal direcionado aos “próprios” neurônios, decorrente das semelhanças eletrofisiológicas entre neurônios e bactérias em termos de gradientes de potencial transmembrana e cargas aniônicas nas macromoléculas da membrana externa. Os produtos de degradação subsequente dos neurônios necróticos provocam liberação adicional de Aβ, levando a um ciclo crônico e autoperpetuador.
O modelo AD2, postula a DA como uma doença autoimune de imunidade inata e não de imunidade adaptativa. Convencionalmente, qualquer lesão não intencional associada à disfunção imune inata é considerada autoinflamatória em vez de autoimune, surgindo principalmente como um efeito não específico de um estado pró-inflamatório. No entanto, como conjecturado em AD2, a DA é uma doença autoimune de imunidade inata porque Aβ liberada como antibacteriano “confunde” os neurônios ao interpretar como não-próprio, perfurando a sua membrana como faria com uma bactéria.
AD2 representa uma conceituação inovadora da DA como uma doença autoimune e autoinflamatória centrada no cérebro da imunidade inata. O modelo AD2 se esforça para harmonizar outras proposições mecanicistas (incluindo proteopatia, sinaptotoxicidade e mitocondriopatia), ao mesmo tempo que reconhece Aβ como um imunopeptídeo fisiologicamente oligomerizante e parte de um processo imunopático muito maior, abrangente e altamente interligado.
Como os processos imunológicos, incluindo a autoimunidade, estão sujeitos à regulação homeostática (modificação farmacológica do metabolismo dos glicocorticóides para controlar doenças autoimunes adaptativas convencionais), é razoável também levantar a hipótese de processos neuroquímicos existentes como reguladores da imunidade inata do cérebro e como uma possível fonte de “moléculas anti-DA”.
No modelo AD2, a DA é considerada como um distúrbio da imunidade inata centrado no cérebro, envolvendo mecanismos autoimunes e autoinflamatórios simultâneos e reconhecendo Aβ como um imunopeptídeo semelhante a citocina fisiologicamente oligomerizante, que é apenas parte de uma conceituação muito maior e abrangente da DA. Dentro deste modelo, o metabolismo dos aminoácidos L-triptofano e L-arginina emergem como reguladores da imunidade inata, apontando assim para novas abordagens diagnósticas e terapêuticas. Diagnosticamente, AD2 sugere que os níveis séricos ou no líquido cefalorraquidiano (LCR) de metabólitos de L-triptofano ou L-arginina podem ser biomarcadores úteis, além de outros marcadores de neuroinflamação.
Para permitir uma estratégia modificadora da DA, a AD2 propõe a regulação da autoimunidade inata por abordagens como a alteração farmacológica do metabolismo do L-triptofano ou da L-arginina através de: (1) análogos moleculares de L-triptofano ou metabólitos de L-arginina, (2) inibidores enzimáticos das vias metabólicas do L-triptofano e/ou L-arginina, (3) manipulação do microbioma do L-triptofano no metabolismo.
A DA destrói inicialmente as conexões neuronais nas regiões da memória e processamento de informações do cérebro, incluindo o hipocampo e o córtex entorrinal, afetando posteriormente as zonas corticais responsáveis pela linguagem, raciocínio e comportamento social. Essas regiões centrais do cérebro permitem redes de modo padrão e mecanismos de integração multissensorial essenciais para comunicações baseadas no cérebro. No nível celular, as regiões centrais do cérebro são ricas em sinapses para facilitar a transmissão de informações e, portanto, requerem quantidades significativas de energia derivada das atividades mitocondriais. Consequentemente, estas regiões são preferencialmente suscetíveis a processos sinaptotóxicos e mitocondriotóxicos (WEIVER, 2023).
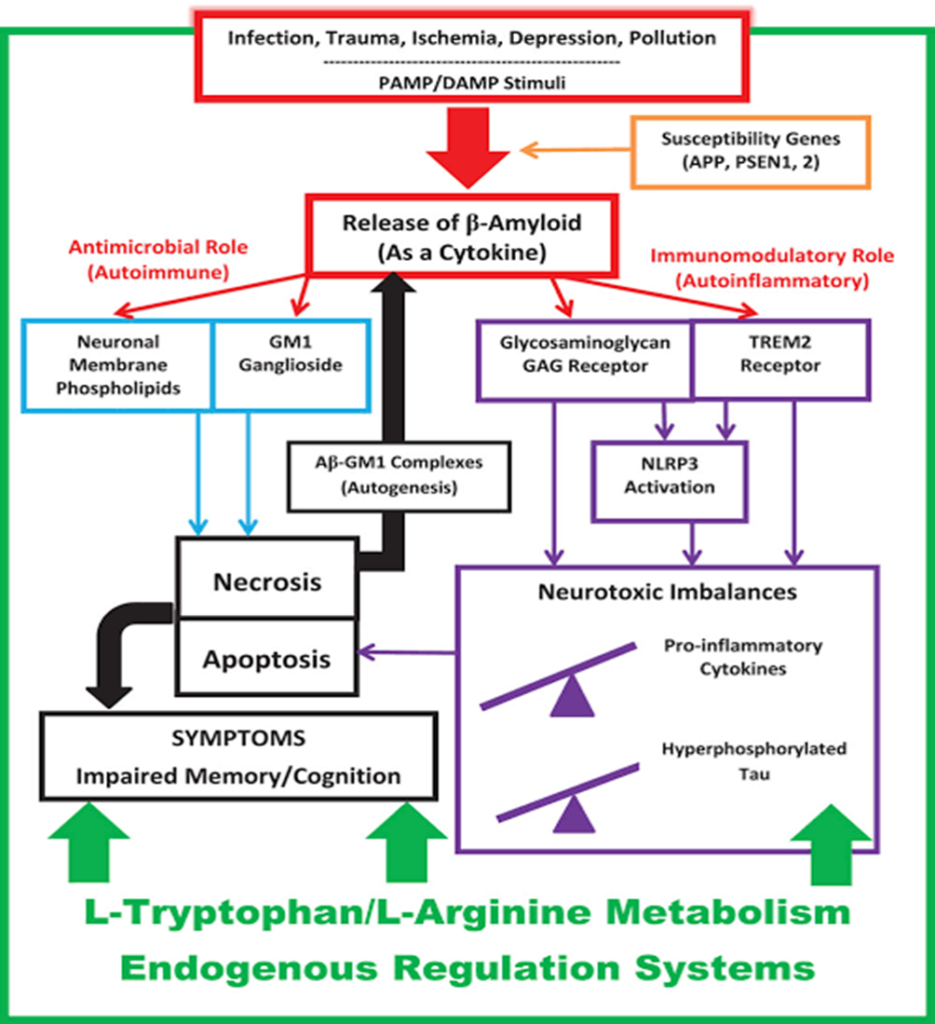
Fonte: Weaver, 2024.
AD2 conceitua a patogênese da DA como emergente de dois processos paralelos: um processo autoimune envolvendo diretamente o Aβ e um processo autoinflamatório que, uma vez iniciado, pode prosseguir independentemente do Aβ para produzir neurotoxicidade por meio de efeitos pró-inflamatórios. Embora o modelo AD2 não possa prever em qualquer pessoa a relação autoimune/autoinflamatória, estes processos paralelos podem ser responsáveis pela associação não linear entre a carga de Aβ e o comprometimento cognitivo, dadas as contribuições variáveis das vias autoimunes e autoinflamatórias para a progressão da DA.
As implicações da dualidade antimicrobiana-imunomoduladora de Aβ, particularmente porque explica outras propostas de mecanismos de doenças, como a sinaptotoxicidade e a tauopatia, para alcançar um modelo amplamente abrangente de DA que unifique múltiplas teorias divergentes em uma única explicação abrangente. Sendo assim, a AD2 destaca a necessidade de pesquisas futuras para avaliar rigorosamente o metabolismo do L-triptofano e da L-arginina, não apenas em termos da base neuroquímica da DA, mas também com o objetivo adicional de identificar novos diagnósticos e terapêuticas que abraçam a noção de que “moléculas anti-DA endógenas” podem constituir uma orientação terapêutica futura. Além disso, o AD2 acentua a necessidade de avaliar continuamente e de forma crítica a capacidade dos modelos de DA em camundongos transgênicos amplamente utilizados e outros bioensaios in vitro/in vivo para analisar com precisão a DA dentro do contexto do modelo AD2. Finalmente, os detalhes do modelo AD2 devem ser explorados para permitir melhores abordagens para a redução de risco em vez de aceitar a generalização abrangente (WEAVER, 2023).
O ponto crucial da AD2 é que Aβ é um imunopeptídeo semelhante a uma citocina que não consegue diferenciar entre bactérias e neurônios, levando a um ataque específico, mas mal direcionado, a si mesmo – isso define a DA como uma resposta autoimune inata. Para validar esta afirmação chave, os próximos passos são a verificação por meio de estudos imuno-histoquímicos de que Aβ possui as propriedades de uma citocina, onde as citocinas são uma ampla categoria estruturalmente diversa de peptídeos de sinalização celular secretados que aux66iliam 6nas interações e comunicações entre células imunocompetentes, sincronizam as respostas do sistema imunológico, influenciam a liberação e ligação ao receptor de outras citocinas e estimula o movimento das células em direção a locais de inflamação, infecção e trauma, funcionando assim como moléculas mensageiras para processos imunológicos inatos e adaptativos; e verificação de que eventos desencadeantes do sistema imunológico, como infecção ou trauma, estimulam a biossíntese e liberação de Aβ.
A verificação através de estudos eletrofisiológicos de que Aβ ataca destrutivamente bactérias e neurônios por mecanismos semelhantes (e correspondentemente que outros AMPs/imunopeptídeos que atacam analogamente neurônios e bactérias de maneira semelhante) e, por último, a verificação de que os neurônios atacados liberam Aβ que se difundem para neurônios adjacentes, desencadeando a liberação contínua de Aβ adicional.
Como os processos imunológicos estão sob forte regulação homeostática, existem sistemas de controle endógenos para a imunidade inata e adaptativa, oferecendo alvos contra doenças relacionadas à disfunção imunológica. Como as doenças autoimunes têm sido historicamente consideradas distúrbios da imunidade adaptativa, os reguladores endógenos têm sido mais bem estudados para esses distúrbios, culminando no uso generalizado de glicocorticóides terapêuticos. Uma via metabólica regulatória análoga precisa ser identificada para a imunidade inata; no contexto da DA, os aminoácidos (particularmente o metabolismo do L-triptofano e da L-arginina) podem cumprir este papel.
Morris (2010) designou a L-arginina como o controlador “mestre e comandante” da imunidade inata. Na DA, existem alterações específicas da região nas concentrações cerebrais de L-arginina e seus metabólitos a jusante (L-citrulina, L-ornitina, agmatina, putrescina, espermidina, espermina, glutamina); correspondentemente, a atividade e a expressão proteica de duas enzimas metabólicas chave da L-arginina, a sintase do óxido nítrico e a arginase, também são alteradas de uma maneira específica da região. Usando análise metabolômica direcionada de metabólitos da via do óxido nítrico/L-arginina na demência
Fleszar et al. (2019) demonstraram uma associação com a patologia, gravidade e alterações estruturais do cérebro. Em um modelo murino de DA, a suplementação de citrulina modificou o declínio cognitivo por meio de alterações nos níveis de L-arginina e óxido nítrico.
Mondanelli et al. (2019) concluíram que as vias metabólicas do L-triptofano e da L-arginina são fundamentais para o controle da imunidade inata e que a interação entre essas duas vias pode ser central para a reprogramação da função das células imunológicas em doenças de base imunológica. Além disso, o L-triptofano e L-arginina demonstraram ser inibidores diretos da oligomerização Aβ (além de outros mecanismos de regulação da imunidade inata), sugerindo a capacidade dos aminoácidos de permitir inibidores multissítios da progressão da DA no contexto do modelo AD2. Estas descobertas demonstram que o metabolismo alterado do L-triptofano e da L-arginina em diversas regiões do cérebro com DA merece uma investigação mais aprofundada para compreender o seu papel na patogênese e/ou progressão da doença.
AD2 modela a DA como uma doença complexa com múltiplas oportunidades de uso medicamentoso, incluindo não apenas a oligomerização de proteínas, mas também de alvos imunológicos (TREM2, NLRP3) e abordagens imunometabólicas (L-triptofano e/ou metabolismo da L-arginina). Tal complexidade, embora seja indiscutivelmente um impedimento a curto prazo, poderá no futuro contribuir para abordagens significativas de medicina personalizada com combinações individualizadas para diferentes pacientes (WEAVER, 2023).
De acordo com Mahajan (2020), a ampla desregulação da transmetilação e síntese/catabolismo de poliaminas, incluindo anormalidades na sinalização de neurotransmissores, ciclo da ureia, metabolismo aspartato-glutamato e síntese de glutationa. Esses resultados implicam alterações no potencial de metilação celular e aumento do fluxo nas vias de transmetilação, aumento da demanda por mecanismos de defesa antioxidante, perturbações no metabolismo intermediário no ciclo da uréia e vias aspartato-glutamato perturbando a bioenergética mitocondrial, aumento da biossíntese e degradação de poliaminas, bem como anormalidades no metabolismo de neurotransmissores relacionados a DA.
A inflamação crônica devido aos distúrbios metabólicos também pode se propagar para o SNC pela entrada de moléculas inflamatórias na área do tronco cerebral, o que pode afetar os núcleos colinérgicos como o tegmento pontino laterodorsal e núcleo tegmental pedunculopontino que têm eferências para o córtex e colinérgicos do prosencéfalo basal (ZOLKIPLI-CUNNINGHAM, 2017). Os resultados, sugerem que a inflamação periférica e central estão correlacionadas ao comprometimento cognitivo e à disfunção colinérgica (CHANG, 2019).
A disfunção metabólica também é outra marca registrada da DA. Como mencionado acima, o GM pode produzir metabólitos bioativos, como SCFAs e neurotransmissores, que podem modular o sistema imunológico e influenciar as atividades cerebrais . Esses metabólitos podem cruzar a BHE para afetar a cognição direta ou indiretamente através de mecanismos imunológicos, neuroendócrinos ou vagais.
Uma meta-análise de estudos observacionais relatou que níveis aumentados de GABA estavam associados a um menor risco de DA. Há uma diferença significativa no nível de metabólitos GM entre pacientes com DA e o grupo controle. Vários metabólitos regulados positivamente pelo GM em pacientes com DA, incluindo o ácido indol-3-pirúvico, um metabólito endógeno do triptofano, e SCFAs, foram correlacionados com comprometimento cognitivo. Curiosamente, os pacientes com DP apresentaram o fenômeno oposto, que é a redução dos SCFAs. Além disso, as concentrações de serotonina no soro e na urina de pacientes com DA foram significativamente mais baixas do que nos controles. A maioria das vias metabólicas associadas aos GM foram previstas com base na análise de sequenciamento do microbioma. Portanto, a validação é essencial para comprovar a relação entre determinada patogênese da GM e da DA (SUN, 2022; WHILEY, 2021).
Em resposta a eventos estimuladores de padrões moleculares associados a patógenos/danos, a Aβ é liberada como uma citocina de resposta precoce, desencadeando uma cascata de imunidade inata na qual Aβ exibe dualidade imunomoduladora/antimicrobiana. No entanto, as propriedades antimicrobianas de Aβ resultam em um ataque mal direcionado aos neurônios, decorrentes das semelhanças eletrofisiológicas entre neurônios e bactérias em termos de gradientes de potencial transmembrana e cargas aniônicas em macromoléculas da membrana externa. Os produtos de degradação subsequentes de neurônios necróticos provocam liberação adicional de Aβ, levando a um ciclo crônico e autoperpetuante. Na DA, o imunometabolismo é um “jogador” de controle central na modulação da autoimunidade da incluindo o Aβ como um importante agente molecular, mas rejeita a “hipótese amilóide”, reconhecendo o Aβ como uma citocina fisiologicamente oligomerizante e parte de uma conceituação imunopática maior da DA (WEIVER, 2024).
Microgliose e neuroinflamação são características proeminentes da DA. A microglia responsiva a doenças atende à sua maior demanda energética reprogramando o metabolismo, especificamente, mudando para favorecer a glicólise em vez da fosforilação oxidativa. Assim, o direcionamento do imunometabolismo microglial pode ser um benefício terapêutico para o tratamento da DA, fornecendo vias imunológicas novas e muitas vezes bem compreendidas e suas ações recentemente reconhecidas na DA (CODOCEDO, 2023).
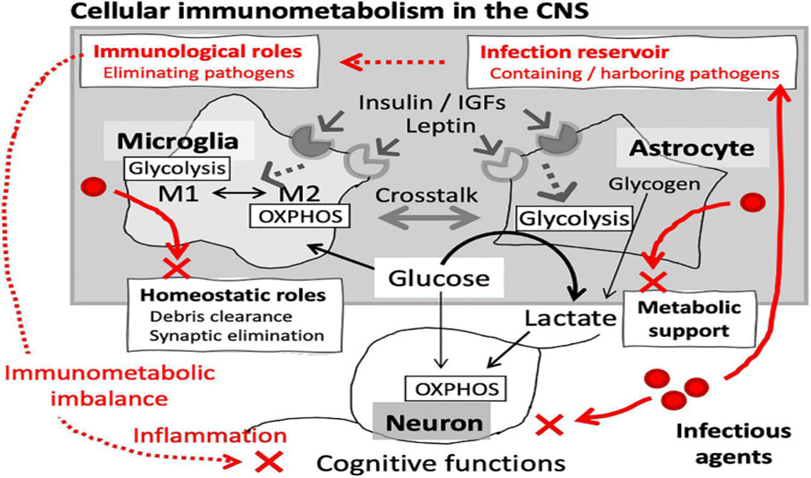
Fonte: Shinjyo, 2021.
Considerando os diversos papéis que essas moléculas desempenham na rede imunometabólica, incluindo o imunometabolismo do SNC é plausível que os danos cerebrais induzidos por infecção possam ser aliviados pela modulação dessas vias de sinalização. Por outro lado, ainda não foi resolvido como as interações glia – patógeno podem impactar o imunometabolismo no SNC e quais os papéis que a leptina e a insulina podem desempenhar nessas interações. Para tanto, as pesquisas são necessárias para compreender os mecanismos subjacentes à inter-relação entre infecção, distúrbios metabólicos e demência (TOO, 2022).
As evidências sugerem que a DA é um distúrbio imunometabólico generalizado no qual o metabolismo celular alterado ocorre nos estágios iniciais da doença. Essa mudança na dependência metabólica não resultou apenas em capacidades glicolíticas cerebrais prejudicadas, mas também em aminoácidos que são essenciais para a reciclagem e síntese de neurotransmissores com comprometimento na fidelidade sináptica, que é a base molecular das funções cognitivas e de memória.
Como perspectiva futura, será interessante compreender como os modificadores do GMB como dieta, geografia, sexo, envelhecimento, exercício e sono, podem dificultar os esforços para direcionar terapeuticamente o GMB. Uma questão importante a ser respondida é se as terapias para DA baseadas no GMB podem ser generalizadas ou se precisam ser personalizadas para o paciente. Para responder adequadamente a esta questão, são necessários ensaios de prevenção e tratamento a longo prazo em humanos, utilizando abordagens terapêuticas baseadas no GMB. Em resumo, embora a investigação que elucida a ligação entre o GMB e a DA tenha evoluído muito em um curto período de tempo, a utilização de novas ferramentas e abordagens irá acelerar o estudo para eventualmente compreender e direcionar terapeuticamente esta ligação.
Ao conceituar Aβ como uma citocina e a DA como uma doença autoimune/autoinflamatória inata, a AD2 oferece um novo paradigma molecular para explicar a cascata precisa de eventos bioquímicos que culminam na DA; por sua vez, esta compreensão biomolecular detalhada facilitará não só uma melhor compreensão da patogênese e dos fatores de risco, mas também conduzirá a melhores diagnósticos e terapêuticas, permitindo a identificação de alvos conhecidos e inexplorados. Portanto, identificar reguladores endógenos dessa autoimunidade e projetar análogos semelhantes a medicamentos representa uma nova abordagem terapêutica para a DA.
Contrariamente à hipótese da cascata amilóide, a proteína Aβ pode ser protetora para os neurônios do SNC nas fases iniciais de eventos de estresse. Por outro viés, quando os estressores crônicos persistem é que a proteína Aβ se torna superexpressa e forma oligômeros e protofibrilas com toxicidade. Portanto, as alterações fisiopatológicas que ocorrem no ambiente do SNC e que induzem estresse neuronal crônico explica a heterogeneidade na trajetória da doença dependendo da combinação de comorbidades subjacentes.
CONSIDERAÇÕES FINAIS
Estudos evidenciaram que a fisiopatologia da DA envolve a deposição da proteína Aβ e a hiperfosforilação da proteína Tau no SNC, as quais são potencializadas por estados inflamatórios (neuroinflamação) capazes de gerar neurodegeneração e progressão da doença. As principais células envolvidas neste processo são a micróglia e os astrócitos no SNC, que a partir da liberação de mediadores inflamatórios geram um processo cíclico de estímulo à neuroinflamação, levando a níveis cada vez maiores de lesão, degeneração e morte neuronal.
Os metabólitos imunológicos específicos do sexo, redes e vias genéticas estão associados à patogênese e progressão da DA. Indivíduos do sexo feminino com DA exibem endofenótipos de imunometabolismo microglial caracterizados por diminuição do metabolismo do glutamato e atividade elevada da via da interleucina-10. Indivíduos do sexo feminino com DA mostraram uma mudança nas comunicações célula-célula mediadas por glutamato entre neurônios excitatórios para microglia e astrócito.
Os resultados apontam para uma nova compreensão da base molecular da predominância feminina na DA, e garantem futuras validações independentes com coortes de pacientes etnicamente diversas para estabelecer uma provável relação causal do imunometabolismo microglial nas diferenças sexuais na DA. Na conclusão dos autores, as diferenças na regulação do imunometabolismo parecem desempenhar um papel importante nas manifestações biológicas específicas do sexo da DA, em particular na forma específica da microglia. Esta nova compreensão poderia ajudar a melhorar o desenvolvimento de terapêuticas, ensaios clínicos e tratamento de pacientes com DA (O’MAHONY, 2024).
Contrariamente à ideia avançada pela hipótese da cascata amilóide, a proteína Aβ pode ser protetora para os neurônios do SNC nas fases iniciais de eventos de estresse. Somente quando os estressores crônicos persistem é que o Aβ se torna superexpresso e forma oligômeros e protofibrilas tóxicos. Portanto, acredita-se que a proteína Aβ não é produzida de forma anormal, mas sim uma molécula de ocorrência normal que faz parte do sistema imunológico do cérebro (FRIZONE, 2022).
O desenvolvimento da DA é acompanhado por uma diminuição na diversidade de espécies de microrganismos comensais levando a um desequilíbrio da microbiota conhecido como disbiose. Este desequilíbrio leva ao aumento da permeabilidade da barreira intestinal, o que prejudica as vias de comunicação do eixo cérebro-intestino, aumentando a neuroinflamação, o que promove o processo de neurodegeneração. Por outro lado, os estudos com diferentes substâncias antioxidantes demonstraram restaurar o eixo cérebro-intestino. Além disso, fornecem informações importantes para definir novos marcadores baseados em microbiomas para apoiar futuras intervenções e ensaios clínicos que podem elucidar as interações entre antioxidantes na tríade microbiota-intestino-cérebro.
Devido as semelhanças impressionantes entre as moléculas de gordura que compõem as membranas das bactérias e as membranas das células cerebrais, a proteína Aβ não consegue distinguir a diferença entre as bactérias invasoras e as células cerebrais hospedeiras e ataca erroneamente as próprias células cerebrais que deveria estar protegendo (MEIER-STEPHENSON, 2022 apud MOURA, 2024a).
A disfunção mitocondrial tem sido associada a patologias metabólicas como diabetes, obesidade, hipertensão, disbiose bem como doenças neurodegenerativas tais como a DA, cerne da presença pesquisa. O distúrbio metabólico é o evento mais precoce no progresso da DA, no qual a disfunção mitocondrial é a principal responsável por isso, uma vez que as mitocôndrias são vitais para o metabolismo neuronal. Durante o processo de envelhecimento, os danos mitocondriais, incluindo a mutação do mtDNA, a redução das atividades das enzimas mitocondriais e o potencial da membrana mitocondrial, irão acumular-se gradualmente (SHARMA, 2021).
Quando o dano excede o limite, os neurônios apresentam distúrbios metabólicos significativos, que é a ocorrência mais precoce da DA. Posteriormente, uma série de distúrbios metabólicos ocorrem nas células devido à energia insuficiente, como autofagia e dobramento incorreto da proteína Aβ. Essas patologias crônicas geralmente começam individualmente e, com o tempo, podem combinar-se com outras modalidades crônicas para aumentar gradativamente o estresse aplicado aos neurônios precedendo os eventos descritos na hipótese da cascata Aβ (WEIDLIN, 2020).
Além disso, a disfunção da autofagia mitocondrial é geralmente a causa da deterioração patológica da DA, entre a qual a disfunção mitocondrial desempenha um papel central na patogênese. A redução de ATP no fornecimento insuficiente de energia celular também inibirá o processo de autofagia mitocondrial que consome energia.
De acordo com a hipótese mitocondrial na DA, as mitocôndrias defeituosas levarão ao aumento de espécies reativas de oxigênio e deposição de Aβ, autofagia suprimida e outras características patológicas relacionadas à DA (REISS, 2022; REDDY, 2019). Assim, a recuperação da função mitocondrial para elevar a geração de energia e ativar na autofagia é considerada uma abordagem potencial para prevenir a progressão da DA. Em pacientes com DA, um grande número de mitocôndrias danificadas é observado sob microscopia eletrônica nos neurônios do SNC.
Os estudos revelam hipometabolismo em regiões do cérebro, como o hipocampo e o córtex cingular posterior, que são particularmente vulneráveis a patologia da DA. Além disso, a disfunção mitocondrial ligada a deficiência de estradiol agrava o estresse oxidativo e prejudica a produção de ATP, o que acaba por afetar a atividade sináptica e a resiliência neuronal. Portanto, não há dúvida de que a disfunção mitocondrial é uma marca registrada na patologia da DA (GORHAM et al., 2023; SOUSA et al., 2023; BARRETO, 2023).
A combinação de múltiplos distúrbios crônicos pode levar ao estresse neuronal ilustrando organicamente as inúmeras possibilidades na heterogeneidade na trajetória da doença. Além disso, a “mistura” da DA com outras patologias crônicas explicaria em parte porque é difícil identificar biomarcadores específicos em biofluídos periféricos, uma vez que os biomarcadores para patologias não relacionadas com a DA mascarariam os marcadores específicos.
As novas evidências sugeriram que a DA é um distúrbio imunometabólico generalizado no qual o metabolismo celular alterado ocorre nos estágios iniciais da doença. Essa mudança na dependência metabólica não resultou apenas em capacidades glicolíticas cerebrais prejudicadas, mas também em aminoácidos que são essenciais para a reciclagem e síntese de neurotransmissores com comprometimento na fidelidade sináptica, que é a base molecular das funções cognitivas e de memória.
Recentemente, a reprogramação metabólica foi identificada como um mediador de alterações relacionadas ao exercício na cognição e nas funções imunológicas, uma vez que o exercício atenuou a expressão de citocinas inflamatórias dependentes da idade e o declínio cognitivo em camundongos, enquanto diminuía as enzimas glicolíticas e aumentava a fagocitose na microglia isolada. No entanto, se o exercício exerce efeitos benéficos ao promover OXPHOS ou FAO, ou o uso de substratos alternativos, ainda precisa ser elucidado. Embora nenhum estudo até o momento tenha investigado se a reprogramação metabólica está subjacente às alterações relacionadas ao exercício na inflamação e na patologia em um contexto específico da DA, esses achados sugerem que o exercício é um caminho promissor para a investigação terapêutica (MELA 2020 apud FAIRLEY et al, 2021).
Evidências emergentes indicam que a função metabólica desempenha um papel crítico na regulação da função microglial na DA, com programação metabólica subjacente a diversas funções e fenótipos imunológicos microgliais. Além disso, estratégias terapêuticas que modulam a programação metabólica microglial demonstraram efeitos neuroprotetores, reduzindo a carga de amiloide e tau e melhorando os déficits cognitivos. Estas descobertas sugerem que a função microglial e o metabolismo são processos intrinsecamente associados na DA, no entanto, são necessárias mais pesquisas para elucidar as vias mecanísticas envolvidas na regulação metabólica na microglia, o que pode, por sua vez, ajudar na identificação de alvos drogáveis para modular o comprometimento imunometabólico.
O modelo intitulado “Alzheimer como Doença Autoimune” (AD2) postula a DA como uma doença autoimune de imunidade inata e não de imunidade adaptativa. Convencionalmente, qualquer lesão não intencional associada à disfunção imune inata é considerada autoinflamatória em vez de autoimune, surgindo principalmente como um efeito não específico de um estado pró-inflamatório. No entanto, como conjecturado em AD2, a DA é uma doença autoimune de imunidade inata porque Aβ liberada como antibacteriano “confunde” os neurônios ao interpretar como não-próprio, perfurando a sua membrana como faria com uma bactéria (WEIVER, 2023).
Tendo em vista essas considerações, sugere-se que o paradigma AD2 representa uma conceituação inovadora da DA como uma doença autoimune e autoinflamatória centrada no cérebro da imunidade inata incluindo proteopatia, sinaptotoxicidade e mitocondriopatia, ao mesmo tempo que reconhece Aβ como um imunopeptídeo fisiologicamente oligomerizante e parte de um processo imunopático muito maior, abrangente e altamente interligado.
REFERÊNCIAS
AGUSTI, A., GARCIA-PARDO, M.P, LOPEZ-ALMELA, I., CAMPILLO, I., MAES, M., ROMANI-PEREZ, M., SANZ, Y. Interação entre o eixo intestino-cérebro, obesidade e função cognitiva. Fronteiras em Neurociências, 2018.
ALFORD, S. et al. Obesity as a risk factor for Alzheimer’s disease: weighing the evidence. Obesity Reviews, v. 19, n. 2, p. 269-280, 2018.
ANDRADE-GUERRERO J, SANTIAGO-BALMASEDA A, JERONIMO-AGUILAR P, VARGAS-RODRÍGUEZ I, CADENA-SUÁREZ AR, SÁNCHEZ-GARIBAY C, POZO-MOLINA G, MÉNDEZ-CATALÁ CF, CARDENAS-AGUAYO MD, DIAZ-CINTRA S, PACHECO-HERRERO M, LUNA-MUÑOZ J, SOTO-ROJAS LO. Alzheimer’s Disease: Na Updated Overview of Its Genetics. Int J Mol Sci. 2023 Feb 13;24(4):3754. Doi: 10.3390/ijms24043754. PMID: 36835161; PMCID: PMC9966419.
ASHRAFI G, SCHLEHE JS, LAVOIE MJ, SCHWARZ TL. A mitofagia de mitocôndrias danificadas ocorre localmente em axônios neuronais distais e requer PINK1 e Parkin. J Cell Biol. 2014;206:655–70.
ASHLEIGH T, SWERDLOW RH, BEAL MF. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement. 2023 Jan;19(1):333-342. doi: 10.1002/alz.12683. Epub 2022 May 6. PMID: 35522844.
ARNOLD, Steven E. et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nature Reviews Neurology. V. 14, n. 3, p. 168–181, 2018.
ARNOLD, Steven E. et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nature Reviews Neurology. v. 14, n. 3, p. 168–181, 2018.
BANO N, KHAN S, AHAMAD S, KANSHANA JS, DAR NJ, KHAN S, NAZIR A, BHAT SA. Microglia and gut microbiota: A double-edged sword in Alzheimer’s disease. Ageing Res Rev. 2024 Nov;101:102515. doi: 10.1016/j.arr.2024.102515. Epub 2024 Sep 24. PMID: 39321881.
BARAGE, SH.; SONAWANE, KD. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides. V. 52, p. 1–18, 2015
BARNES LL, WILSON RS, BIENIAS JL, SCHNEIDER JÁ, EVANS DA, BENNETT DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry. 2005 Jun;62(6):685-91. Doi: 10.1001/archpsyc.62.6.685. PMID: 15939846.
BUSCHE, Marc Aurel; HYMAN, Bradley T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nature Neuroscience. V. 23, n. 10, p. 1183–1193, 2020.
BUTTERFIELD, D. Allan; HALLIWELL, Barry. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nature Reviews Neuroscience. V. 20, n. 3, p.148–160, 2019.
BUTTERFIELD, D. A.; LAUDERBACK, C. M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide- associated free radical oxidative stress. Free Radic. Biol. Med., v. 32, n. 11, p. 1050-1060, 2002.
BUTTERFIELD, D. A; HALLIWELL, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nature Reviews Neuroscience. V. 20, n. 3, p. 148–160, 2019.
BIANCHI VE. Impact of Testosterone on Alzheimer’s Disease. World J Mens Health. 2025:243-256.
BUGLIO DS, MARTON LT, LAURINDO LF, GUIGUER EL, ARAÚJO AC, BUCHAIM RL, GOULART RA, RUBIRA CJ, BARBALHO SM. The Role of Resveratrol in Mild Cognitive Impairment and Alzheimer’s Disease: A Systematic Review. J Med Food. 2022 Aug;25(8):797-806. Doi: 10.1089/jmf.2021.0084. Epub 2022 Mar 28. PMID: 35353606.
BUTTERFIELD, D. A; HALLIWELL, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nature Reviews Neuroscience. V. 20, n. 3, p. 148–160, 2019.
BUTTERFIELD DA, PERLUIGI M, SULTANA R. Oxidative stress in Alzheimer’s disease brain: new insights from redox proteomics. Eur J Pharmacol. 2006 Sep 1;545(1):39-50. Doi: 10.1016/j.ejphar.2006.06.026. Epub 2006 Jun 15. PMID: 16860790.
CAO W, ZHENG H. Sistema imunológico periférico no envelhecimento e na doença de Alzheimer. Mol Neurodegener 13:58, 2018.
CECI C, LACAL PM, BARBACCIA ML, MERCURI NB, GRAZIANI G, LEDONNE A. The VEGFs/VEGFRs system in Alzheimer’s and Parkinson’s diseases: Pathophysiological roles and therapeutic implications. Pharmacol Res. 2024 Mar;201:107101. Doi: 10.1016/j.phrs.2024.107101. Epub 2024 Feb 7. PMID: 38336311.
CHOPRA et al., Curcumin Nanoparticles as Therapeutic Agents for Drug Targets. Molecules, 26(16):4998, 2021.
COHEN M.V. Free radicals in ischemic and reperfusion myocardial injury: is this time for clinical trials? Annals of Internal Medicine. 1989, 111: 918-931.
COLACURCIO, D. J. & NIXON, R. A. Disorders of lysosomal acidification—the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Ver. 32, 75–88 (2016).
CHANDRA S, SISODIA SS, VASSAR RJ. The gut microbiome in Alzheimer’s disease: what we know and what remains to be explored. Mol Neurodegener. 2023 Feb 1;18(1):9. Doi: 10.1186/s13024-023-00595-7. PMID: 36721148; PMCID: PMC9889249.
CHAUHAN, V.; CHAUHAN, A. Oxidative stress in alzheimer’s disease. Pathophysiology, v. 13, n. 3, p. 195-208, 2006.
CRYAN, J. F.; O’RIORDAN, K.J., COWAN, C. S. M.The Microbiota-Gut-Brain Axis. PhysiolRev, v. 99, n.: 4, p.1877–2013, 2019. CUMMINGS J, BENSON D. Subcortical Dementia. Arch Neurol 41: 874-879, 1984.
CUMMINS N, TWEEDIE A, ZURYN S, BERTRAN-GONZALEZ J, GÖTZ J. Tau associado à doença prejudica a mitofagia ao inibir a translocação de Parkin para as mitocôndrias. The EMBO J. 2019;38:e99360.
CUMMINGS J, OSSE AML, CAMMANN D, POWELL J, CHEN J. Anti-Amyloid Monoclonal Antibodies for the Treatment of Alzheimer’s Disease. BioDrugs. 2024 Jan;38(1):5-22. doi: 10.1007/s40259-023-00633-2. Epub 2023 Nov 13. PMID: 37955845; PMCID: PMC10789674.
CUMMINGS JL, BENSOM DF. The role of the nucleus basalis of Meinert im dementia: Review ande reconsideration. Alzheimer’s Dis Assoc Disorde 1987; 1: 128-145.
CHAPLYGINA A, ZHDANOVA D. Mitochondrial Fragmentation as a Key Driver of Neurodegenerative Disease. Curr Alzheimer Res. 2025 Jan 8. Doi: 10.2174/0115672050366194250107050650. Epub ahead of print. PMID: 39791144.
CHEN H, ZENG Y, WANG D, LI Y, XING J, ZENG Y, LIU Z, ZHOU X, Fan H. Neuroinflammation of Microglial Regulation in Alzheimer’s Disease: Therapeutic Approaches. Molecules. 2024 Mar 26;29(7):1478. Doi: 10.3390/molecules29071478. PMID: 38611758; PMCID: PMC11013124.
CHEN TB, YIAO SY, SUN Y, LEE HJ, YANG SC, CHIU MJ, et al. (2017). Comorbidity and dementia: A nationwide survey in Taiwan. PLoS One, 12:e0175475.
CHEN H, MCCAFFERY JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007 Aug 10;130(3):548-62. Doi: 10.1016/j.cell.2007.06.026. PMID: 17693261.
CODOCEDO JF, MERA-REINA C, LIN PB, PUNTAMBEKAR SS, CASALI BT, JURY N, MARTINEZ P, LASAGNA-REEVES CA, LANDRETH GE. Therapeutic targeting of immunometabolism in Alzheimer’s disease reveals a critical reliance on Hexokinase 2 dosage on microglial activation and disease progression. bioRxiv, 2023 Nov 15:2023.11.11.566270. doi: 10.1101/2023.11.11.566270. Update in: Cell Rep. 2024 Jul 23;43(7):114488. doi: 10.1016/j.celrep.2024.114488. PMID: 38014106; PMCID: PMC10680613.
COSTA, M.R. Switch of innate to adaptative immune responses in the brain of patients with Alzheimer’s disease correlates with tauopathy progression. Npj Aging 10, 19 (2024).
CRYAN, J. F.; O’RIORDAN, K.J., COWAN, C. S. M.The Microbiota-Gut-Brain Axis. PhysiolRev, v. 99, n.: 4, p.1877–2013, 2019.
CUI J, SHEN Y, LI R. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med. 2013;19:197–209.
CUMMINGS J, LEE G, RITTER A, SABBAGH M, ZHONG K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement (N Y). 2019 Jul 9;5:272-293. Doi: 10.1016/j.trci.2019.05.008. PMID: 31334330; PMCID: PMC6617248.
DICKSON DW Neuropathological diagnosis of Alzheimer’s disease: a perspective from longitudinal clinicopathological studies. Neurobiol Aging, 1997. 18:21−26.
DE I, MAKLAKOVA V, LITSCHER S, BOYD MM, KLEMM LC, WANG Z, et al. Microglial responses to CSF1 overexpression do not promote the expansion of other glial lineages. J Neuroinflamm. 2021;18(1):162.
FAIRLEY LH, WONG JH, BARRON AM. Mitochondrial Regulation of Microglial Immunometabolism in Alzheimer’s Disease. Front Immunol. 2021 Feb 25;12:624538. Doi: 10.3389/fimmu.2021.624538. PMID: 33717134; PMCID: PMC7947196.
FALCO A. Doença de Alzheimer: Hipóteses Etiológicas e Perspectivas de Tratamento. Quim. Nova, Vol. 39, No. 1, 63-80, 2016.
FANG EF, HOU Y, PALIKARAS K, ADRIAANSE BA, KERR JS, YANG B, et al. A mitofagia inibe a patologia amiloide-β e tau e reverte déficits cognitivos em modelos de doença de Alzheimer. Nat Neurosci. 2019;22:401–12.
FERREIRA JCB. Mitochondrial Dysfunction in Degenerative Diseases. Cells. 2022 May 5;11(9):1546. doi: 10.3390/cells11091546. PMID: 35563852; PMCID: PMC9103981.
FERREIRA A.L.A. & MATSUBARA 1997. L.S. Radicais livres: conceitos, doenças relacionadas, sistema de defesa e estresse Oxidativo. Revista da Associação Médica Brasileira. 43: 61-68.
FORNY-GERMANO, L. et al. The Role of Leptin and Adiponectin in Obesity Associated Cognitive Decline and Alzheimer‟s Disease. Frontiers in Neuroscience, v. 12, 2018.
GARCIA-ESCUDERO V., MARTIN-MAESTRO P., PERRY G., AVILA J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell. Longev. 2013;2013:162152.
GAUGLER JE, HOBDAY JV, ROBBINS JC, BARCLAY MP. CARES(®) Dementia Care for Families™: Effects of Online, Psychoeducational Training on Knowledge of Person-Centered Care and Satisfaction. J Gerontol Nurs, 2015.
GILLETT MJ, MARTINS RN, CLARNETTE RM, CHUBB AS, BRUCE DG, YEAP BB. Relationship between testosterone, sex hormone binding globulin and plasma amyloid beta peptide 40 in older men with subjective memory loss or dementia. J Alzheimers Dis. 2023;5:267–269.
GUSTAVSSON A, NORTON N, FAST T, FRÖLICH L, GEORGES J, HOLZAPFEL D, KIRABALI T, KROLAK-SALMON P, ROSSINI PM, FERRETTI MT, LANMAN L, CHADHA AS, VAN DER FLIER WM. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimers Dement. 2023 Feb;19(2):658-670. Doi: 10.1002/alz.12694. Epub 2022 Jun 2. PMID: 35652476.
GUAN D, LIANG C, ZHENG D, LIU S, LUO J, CAI Z, ZHANG H, CHEN J. The role of mitochondrial remodeling in neurodegenerative diseases. Neurochem Int. 2025 Jan 9;183:105927. doi: 10.1016/j.neuint.2024.105927. Epub ahead of print. PMID: 39798853.
GUTTERIDGE, J.M.C. Lipid peroxidation and antioxidants as biomarkers of tissues damage. Clin.Chem., v.41, p.1819-1828, 1995.
HARDY J, SELKOE DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002; 297: 353-56.
HAAKE, A et al. Na update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opinion on Drug Safety. V. 19, n. 2, p. 147–157, 2020.
HALLIWELL B, Gutteridge J. Free Radicals in Biology and Medicine. 4th edn. New York: Oxford University Press Inc; 2007.
HALLIWELL B. Lipid peroxidation, antioxidants and cardiovascular disease: how should we move forward ? Cardiovas.Res., v. 47, p.410-418, 2000.
HE C, SUMPTER R JR, LEVINE B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy. 2012 Oct;8(10):1548-51. Doi: 10.4161/auto.21327. Epub 2012 Aug 15. PMID: 22892563; PMCID: PMC3463459.
HENEKA MT, CARSON MJ, EL KHOURY J, LANDRETH GE, BROSSERON F, FEINSTEIN DL, JACOBS AH, WYSS-CORAY T, VITORICA J, RANSOHOFF RM, HERRUP K, FRAUTSCHY SA, FINSEN B, BROWN GC, VERKHRATSKY A, YAMANAKA K, KOISTINAHO J, LATZ E, HALLE A, PETZOLD GC, TOWN T, MORGAN D, SHINOHARA ML, PERRY VH, HOLMES C, BAZAN NG, BROOKS DJ, HUNOT S, JOSEPH B, DEIGENDESCH N, GARASCHUK O, BODDEKE E, DINARELLO CA, BREITNER JC, COLE GM, GOLENBOCK DT, KUMMER MP. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015 Apr;14(4):388-405. doi: 10.1016/S1474-4422(15)70016-5. PMID: 25792098; PMCID: PMC5909703.
HEO JH, HYON-LEE, LEE KM. The possible role of antioxidant vitamin C in Alzheimer’s disease treatment and prevention. Am J Alzheimers Dis Other Demen. 2023 Mar;28(2):120-5. Doi: 10.1177/1533317512473193. Epub 2023 Jan 9. PMID: 23307795; PMCID: PMC10852723.
HOOGMARTENS, J; CACACE, R; BROECKHOVEN, C V. Insight intothe genetic etiology of Alzheimer’s disease: A comprehensive review of the role of rarevariants. Alzheimer’s Dement. V. 13, n. 1, 2021.
HOU Y, CALDWELL JZK, LATHIA JD, LEVERENZ JB, PIEPER AA, CUMMINGS J, CHENG F. Microglial immunometabolism endophenotypes contribute to sex difference in Alzheimer’s disease. Alzheimers Dement. 2024 Feb;20(2):1334-1349. Doi: 10.1002/alz.13546. Epub 2023 Nov 20. PMID: 37985399; PMCID: PMC10916937.
HU Y, CAO K, WANG F, WU W, MAI W, QIU L, LUO Y, GE WP, SUN B, SHI L, ZHU J, ZHANG J, WU Z, XIE Y, DUAN S, GAO Z. Dual roles of hexokinase 2 in shaping microglial function by gating glycolytic flux and mitochondrial activity. Nat Metab. 2022 Dec;4(12):1756-1774. doi: 10.1038/s42255-022-00707-5. Epub 2022 Dec 19. PMID: 36536134.
HU YT, BOONSTRA J, MCGURRAN H, STORMMESAND J, SLUITER A, BALESAR R, VERWER R, SWAAB D, BAO AM. Sex differences in the neuropathological hallmarks of Alzheimer’s disease: focus on cognitively intact elderly individuals. Neuropathol Appl Neurobiol. 2021 Dec;47(7):958-966. Doi: 10.1111/nan.12729. Epub 2021 May 27. PMID: 33969531; PMCID: PMC9290663.
HU, X.; WANG, T.; JIN, F. Alzheimer’s disease and gut microbiota. Science China Life Sciences, v.59, n.10, p.1006-1023, 2019.
HUANG, T. RAFAEL GUIMARÃES, N. TODD, C. FERGUSON, K.M. WEISS, F.R. STAUFFER, B. MCDERMOTT, B.T. HURTLE, T. SAITO, T.C. SAIDO, M.L. MACDONALD, G.E. HOMANICS, A. THATHIAH, G protein–biased GPR3 signaling ameliorates amyloid pathology in a preclinical Alzheimer’s disease mouse model, Proc. Natl. Acad. Sci. U.S.A.119 (40) e2204828119, https://doi.org/10.1073/pnas.2204828119 (2022).
HUNG COY, LIVESEY FJ. O processamento alterado da γ-secretase de APP interrompe a função do lisossomo e do autofagossomo na doença de Alzheimer monogênica. Cell Rep. 2018;25:3647–60.e2.
KANDIMALLA, Ramesh; THIRUMALA, Vani; REDDY, P. Hemachandra. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochimica et Biophysica Acta – Molecular Basis of Disease. V. 1863, n. 5, p. 1078–1089, 2017.0.
LEE S, BYUN MS, Yi D, et al. Plasma Leptin and Alzheimer Protein Pathologies Among Older Adults. JAMA Netw Open, 7(5) 249539, 2024.
LEE, JH., YANG, DS., GOULBOURNE, C.N. et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci 25, 688–701, 2022.
LEE SE, KWON D, SHIN N, KONG D, KIM NG, KIM HY, et al. Acúmulo de APP-CTF induz disfunção de mitofagia no modelo iNSCs da doença de Alzheimer. Cell Death Discov. 2022;8:1.
LEE SI, JEONG W, LIM H, CHO S, LEE H, JANG Y, et al. Astrócitos humanos portadores de APOE4 fornecem colesterol em excesso para promover a expansão da balsa lipídica neuronal e a geração de Aβ. Stem cell Rep. 2021;16:2128–37.
LEPIARZ-RABA, I., GBADAMOSI, I., FLOREA, R. et al. Regulação metabólica da fagocitose microglial: Implicações para a terapêutica da doença de Alzheimer. Transl Neurodegener 12 , 48 (2023).
LEFTEROV I, WOLFE CM, FITZ NF, NAM KN, LETRONNE F, BIEDRZYCKI RJ, et al. Diferenças orquestradas por APOE2 em perfis transcriptômicos e lipidômicos de cérebro com DA post-mortem. Alzheimers Res Ther. 2019;11:113.
LIEBERMAN O. The synaptic autophagy cycle. J. Mol. Biol., 432 (2020), p. 2589-2604
LLANOS-GONZÁLEZ E, HENARES-CHAVARINO ÁA, PEDRERO-PRIETO CM, GARCÍA-CARPINTERO S, FRONTIÑÁN-RUBIO J, SANCHO-BIELSA FJ, et al. Interplay Between Mitochondrial Oxidative Disorders and Proteostasis in Alzheimer’s Disease. Frontiers in Neuroscience. 2020;13:1444.
LIANG J, WANG C, ZHANG H, HUANG J, XIE J, CHEN N. Exercise-Induced Benefits for Alzheimer’s Disease by Stimulating Mitophagy and Improving Mitochondrial Function. Front Aging Neurosci. 2021 Oct 1;13:755665. DOI: 10.3389/FNAGI.2021.755665. PMID: 34658846; PMCID: PMC8519401.
LIVINGSTON G, HUNTLEY J, LIU KY, COSTAFREDA SG, SELBÆK G, ALLADI S, AMES D, BANERJEE S, BURNS A, BRAYNE C, FOX NC, FERRI CP, GITLIN LN, HOWARD R, KALES HC, KIVIMÄKI M, LARSON EB, NAKASUJJA N, ROCKWOOD K, SAMUS Q, SHIRAI K, SINGH-MANOUX A, SCHNEIDER LS, WALSH S, YAO Y, SOMMERLAD A, MUKADAM N. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet. 2024 Aug 10;404(10452):572-628. Doi: 10.1016/S0140-6736(24)01296-0. Epub 2024 Jul 31. PMID: 39096926.
MARTÍN-MAESTRO P, GARGINI R, A. SPROUL A, GARCÍA E, ANTÓN LC, NOGGLE S, et al. Mitophagy failure in fibroblasts and iPSC-derived neurons of Alzheimer’s Disease-associated presenilin 1 Mutation. Front Mol Neurosci. 2017;10:291.
MCCARTY M.F., LERNER A., DINICOLANTONIO J.J., ILOKI-ASSANGA S.B. High Intakes of Bioavailable Phosphate May Promote Systemic Oxidative Stress and Vascular Calcification by Boosting Mitochondrial Membrane Potential-Is Good Magnesium Status an Antidote? Cells. 2021;10:1744.
MAINARDI M, SPINELLI M, SCALA F, MATTERA A, FUSCO S, D’ASCENZO M, GRASSI C. Loss of Leptin-Induced Modulation of Hippocampal Synaptic Trasmission and Signal Transduction in High-Fat Diet-Fed Mice. Front Cell Neurosci. 2017 Jul 28;11:225. Doi: 10.3389/fncel.2017.00225. PMID: 28804449; PMCID: PMC5532388.
MAHAJAN UV, VARMA VR, GRISWOLD ME, BLACKSHEAR CT, NA Y, OOMMEN AM, VARMA S, TRONCOSO JC, PLETNIKOVA O, O’BRIEN R, HOHMAN TJ, LEGIDO-QUIGLEY C, THAMBISETTY M. Dysregulation of multiple metabolic networks related to brain transmethylation and polyamine pathways in Alzheimer disease: A targeted metabolomic and transcriptomic study. PLoS Med. 2020 Jan 24;17(1):e1003012. Doi: 10.1371/journal.pmed.1003012. Erratum in: PLoS Med. 2020 Oct 21;17(10):e1003439. Doi: 10.1371/journal.pmed.1003439. PMID: 31978055; PMCID: PMC6980402.
MAIOLI, S. et al. Alterations in brain leptin signalling in spite of unchanged CSF leptin levels in Alzheimer’s disease. Aging Cell, v. 14, n. 1, p. 122-129, 2015.
MARY, A., EYSERT, F., CHECLER, F. et al. Mitofagia na doença de Alzheimer: defeitos moleculares e abordagens terapêuticas. Mol Psychiatry 28 , 202–216 (2023).
MARTORANA, A. et al. Immunosenescence, inflammation and Alzheimer‟s disease. Longevity & Healthspan, v. 1, n. 1, p. 8, 2012.
MARWARHA, G.; GHRIBI, O. Leptin signaling and Alzheimer‟s disease. American Journal of Neurodegenerative Disease, v. 1, n. 3, p. 245, 2012.
MELA V, MOTA BC, MILNER M, MCGINLEY A, MILLS KHG, KELLY ÁM, LYNCH MA. Exercise-induced re-programming of age-related metabolic changes in microglia is accompanied by a reduction in senescent cells. Brain Behav Immun. 2020 Jul;87:413-428. Doi: 10.1016/j.bbi.2020.01.012. Epub 2020 Jan 21. PMID: 31978523.
MEYER MR, KIRMESS KM, EASTWOOD S, WENTE-ROTH TL, IRVIN F, HOLUBASCH MS, VENKATESH V, FOGELMAN I, MONANE M, HANNA L, RABINOVICI GD, SIEGEL BA, WHITMER RA, APGAR C, BATEMAN RJ, HOLTZMAN DM, IRIZARRY M, VERBEL D, SACHDEV P, ITO S, CONTOIS J, YARASHESKI KE, BRAUNSTEIN JB, VERGHESE PB, West T. Clinical validation of the PrecivityAD2 blood test: A mass spectrometry-based test with algorithm combining %p-tau217 and Aβ42/40 ratio to identify presence of brain amyloid. Alzheimers Dement. 2024 May;20(5):3179-3192. doi: 10.1002/alz.13764. Epub 2024 Mar 16. PMID: 38491912; PMCID: PMC11095426.
MOLLER HJ, GRAEBER MB (1998) The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Eur Arch Psychiatry Clin Neurosci 248:111–22.
MORAES-VIEIRA, PMM.; SAGHATELIAN, A; KAHN, BB. Glut4 Expression in Adipocytes Regulates de Novo Lipogenesis and Levels of a Novel Class of Lipids with Anti-Diabetic and Anti-inflammatory Effects. Diabetes (New York, N.Y.), 2016.
MEIER‐STEPHENSON FS, MEIER‐STEPHENSON VC, CARTER MD, et al. Alzheimer’s disease as an autoimmune disorder of innate immunity endogenously modulated by tryptophan metabolites. Alzheimer’s Dement. 2022.
MENZIES, F. M. et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034 (2017).
MOURA, RMBL Correlação do imunometabolismo na doença de alzheimer: hipótese etiológica e tratamentos coadjuvantes com ação antioxidante. Revista Fisio&Terapia, Ciências da Saúde, Medicina, V 28, Ed 139, 2024. Doi: 10.69849/revistaft/th102410242256.
MOURA, RMBL. Disfunção Lisossomal e Mitocondrial do Sistema Imunológico: Hipótese Etiológica da Doença de Alzheimer. Revista Fisio&Terapia, Ciências da Saúde, Medicina. 28, p. 97, 2024a.
MOURA, RMBL; COSTA, C.A.S. Alzheimer´s Disease: Epidemiological Aspects of the use in the treatment of Memantine. REB: Revista Eletrônica de Biologia da PUCSP. 167-174, 2014b.
MOURA, RMBL. Considerações Genéticas sobre a Doença de Alzheimer. Revista Razão e Fé. Pelotas: EDUCAT, v.12, p 93-100, 2010c.
MOUSSA N, DAYOUB N. Exploring the role of COX-2 in Alzheimer’s disease: Potential therapeutic implications of COX-2 inhibitors. Saudi Pharm J. 2023 Sep;31(9):101729. Doi: 10.1016/j.jsps.2023.101729. Epub 2023 Aug 7. PMID: 37638222; PMCID: PMC10448476.
NAFAR F, CLARKE JP, MEAROW KM. Coconut oil protects cortical neurons from amyloid beta toxicity by enhancing signaling of cell survival pathways. Neurochem Int. 2017 Mai;105:64-79.
NGUYEN NM, CHO J, LEE C. Gut Microbiota and Alzheimer’s Disease: How to Study and Apply Their Relationship. Int J Mol Sci. 2023 Feb 17;24(4):4047. Doi: 10.3390/ijms24044047. PMID: 36835459; PMCID: PMC9958597.
NASCIMENTO C et al. Zinc deficiency in alzheimer’s disease: a cross-sectional study with a control group. Revista Brasileira de Geriatria e Gerontologia., 2023.
NAUFEL MF, TRUZZI GM, FERREIRA CM, COELHO FMS. The brain-gut-microbiota axis in the treatment of neurologic and psychiatric disorders. Arq Neuropsiquiatr. 2023 Jul;81(7):670-684. Doi: 10.1055/s-0043-1767818. Epub 2023 Jul 4. PMID: 37402401; PMCID: PMC10371417.
NG, TK et al. Decreased serum brain-derived neurotrophic factor (BDNF) levels in patients with Alzheimer’s disease (AD): A systematic review andmeta-analysis. International Journal of Molecular Sciences. V. 20, n. 2, p. 1–26, 2019.
NITRINI, R. Caramelli P, Bottino CM, Damasceno BP, Bru-cki SM, Anghinah R; Academia Brasileira de Neurologia. Diagnóstico de doença de Alzheimer no Brasil: critérios diagnósticos e exames complementares. Arq Neuropsiquiatria, 2005; 63:713-719.
NIXON, R. A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 120, 4081–4091 (2007).
OCAÑAS SR, PHAM KD, COX JEJ, KECK AW, KO S, AMPADU FA, PORTER HL, ANSERE VA, KULPA A, KELLOGG CM, MACHALINSKI AH, THOMAS MA, WRIGHT Z, CHUCAIR-ELLIOTT AJ, FREEMAN WM. Microglial senescence contributes to female-biased neuroinflammation in the aging mouse hippocampus: implications for Alzheimer’s disease. J Neuroinflammation. 2023, 16;20(1):188.
OERTEL W, SCHULZ JB. Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J Neurochem. 2016 Oct;139 Suppl 1:325-337. Doi: 10.1111/jnc.13750. Epub 2016 Aug 30. PMID: 27577098.
OH, S.J., LEE, HJ., JEONG, Y.J. et al. Evaluation of the neuroprotective effect of taurine in Alzheimer’s disease using functional molecular imaging. Sci Rep 10, 15551 (2020). https://doi.org/10.1038/s41598-020-72755-4.
OSHIMA Y, VERHOEVEN N, CARTIER E, KARBOWSKI M. The OMM-severed and IMM-ubiquitinated mitochondria are intermediates of mitochondrial proteotoxicity-induced autophagy in PRKN/parkin-deficient cells. Autophagy. 2021 Nov;17(11):3884-3886. doi: 10.1080/15548627.2021.1964887. Epub 2021 Sep 5. PMID: 34486484; PMCID: PMC8632337.
O’MAHONY C, HIDALGO-LANUSSA O, Barreto GE. Unveiling FOXO3’s metabolic contribution to menopause and Alzheimer’s disease. Exp Gerontol. 2025 Jan 9;200:112679. Doi: 10.1016/j.exger.2025.112679. Epub ahead of print. PMID: 39778695.
O’MAHONY, S.M; , CLARKE, G; BORRE, Y.E; DINAN, T.G; CRYAN, J.F . Serotonina, metabolismo do triptofano e eixo cérebro-intestino-microbioma Behav Brain Res. 2015; 277: 32-48.
ONYANGO IG, JAUREGUI GV, CARNÁ M, BENNETT JP, STOKIN GB. Neuroinflammation in Alzheimer’s Disease. Biomedicines 9(5): 524, 2021.
ORRENIUS S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Ver Pharmacol Toxicol 2007;47:143-83.
OSSE AML, PANDEY RS, WIRT RA, ORTIZ AA, SALAZAR A, KIMMICH M, TOLEDANO STROM EM, OBLAK A, LAMB B, HYMAN JM, CARTER GW, KINNEY J. Reduction in GABAB on glia induce Alzheimer’s disease related changes. Brain Behav Immun. 2023 May;110:260-275. Doi: 10.1016/j.bbi.2023.03.002. Epub 2023 Mar 9. PMID: 36906075; PMCID: PMC10115139.
PALLADINO MA, BAHJAT FR, THEODORAKIS EA, MOLDAWER LL (2003) Anti-TNF- alpha therapies: the next generation. Nat Ver Drug Discov 2:736-46.
PAKPIAN N, PHOPIN K, KITIDEE K, GOVITRAPONG P, WONGCHITRAT P. Alterações em genes relacionados à dinâmica mitocondrial no sangue periférico de pacientes com doença de Alzheimer. Curr Alzheimer Res. 2020;17:616–25.
PARSONS, C.G.; GRUNER, R.; ROZENTAL, J. et al. – Patch clamp studies on the kinetics and selectivity of N-methyl-D-aspartate receptor antagonism by memantine (1-amino-3,5-dimethyladamantan). Neuropharmacology 1993;32(12):1337-50.
PAUL S, SAHA D, Bk B (2021). Mitochondrial Dysfunction and Mitophagy Closely Cooperate in Neurological Deficits Associated with Alzheimer’s Disease and Type 2 Diabetes. Mol Neurobiol.
PEREZ-GONZALEZ R., ALVIRA-BOTERO M. X., ROBAYO O., ANTEQUERA D., GARZON M., Martín-Moreno A. M., et al. (2014). Leptin gene therapy attenuates neuronal damages evoked by amyloid-β and rescues memory deficits in APP/PS1 mice. Gene Ther. 21 298–308. 10.1038/gt.2013.85.
PETEK B, HÄBEL H, XU H, VILLA-LOPEZ M, KALAR I, HOANG MT, MAIOLI S, PEREIRA JB, MOSTAFAEI S, WINBLAD B, GREGORIC KRAMBERGER M, ERIKSDOTTER M, GARCIA-PTACEK S. Statins and cognitive decline in patients with Alzheimer’s and mixed dementia: a longitudinal registry-based cohort study. Alzheimers Res Ther. 2023 Dec 20;15(1):220. Doi: 10.1186/s13195-023-01360-0. PMID: 38115091; PMCID: PMC10731754.
PETRALIA, M.C.; BATTAGLIA, G.; BRUNO, V.; PENNISI, M.; MANGANO, K.; LOMBARDO, S.D.; FAGONE, P.; CAVALLI, E.; SARACENO, A.; NICOLETTI, F.; et al. The Role of Macrophage Migration Inhibitory Factor in Alzheimer Disease: Conventionally Pathogenetic or Unconventionally Protective? Molecules 2020, 25, 291.
PHILIP, A; WHITE, ND. Gluten, Inflammation, and Neurodegeneration. American Journal Of Lifestyle Medicine , [S.L.], v. 16, n. 1, p. 32-35, jan. 2022.
PIMENOVA AA, HERBINET M, GUPTA I, MACHLOVI SI, BOWLES KR, MARCORA E, GOATE AM. Alzheimer’s-associated PU.1 expression levels regulate microglial inflammatory response. Neurobiol Dis. 2021 Jan;148:105217. Doi: 10.1016/j.nbd.2020.105217. Epub 2020 Dec 8. PMID: 33301878; PMCID: PMC7808757.
PIRES M, REGO AC. Apoe4 and Alzheimer’s Disease Pathogenesis-Mitochondrial Deregulation and Targeted Therapeutic Strategies. Int J Mol Sci. 2023 Jan 1;24(1):778. Doi: 10.3390/ijms24010778. PMID: 36614219; PMCID: PMC9821307.
PISTOLLATO, F. et al. “Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease.” Nutrition reviews v.74, n. 10, p. 624-634, 2016.
PROKIC I, COWLING BS, LAPORTE J. 2014. Amphiphysin 2 (BIN1) in physiology and diseases. J. Mol. Med. 92, 453-463.
PULINA MV, HOPKINS M, HAROUTUNIAN V, GREENGARD P, BUSTOS V. C99 acumula-se seletivamente em neurônios vulneráveis na doença de Alzheimer. Alzheimer’s Dement : J Alzheimer’s Assoc. 2020;16:273–82.
SHARMA C., KIM S., NAM Y., JUNG U.J., KIM S.R. Mitochondrial dysfunction as a driver of cognitive impairment in Alzheimer’s disease. Int. J. Mol. Sci. 2021;22:4850.
SHIPPY DC, OLIAI SF, ULLAND TK. Zinc utilization by microglia in Alzheimer’s disease. J Biol Chem. 2024 May;300(5):107306. Doi: 10.1016/j.jbc.2024.107306. Epub 2024 Apr 20. PMID: 38648940; PMCID: PMC11103939.
SHIPPY DC, ULLAND TK. Microglial Immunometabolism in Alzheimer’s Disease. Front Cell Neurosci 14: 563446, 2020.
SILVA, M.V.F.; LOURES, C.M.G.; ALVES, L.C.V.; DE SOUZA, L.C.; BORGES, K.B.G.; CARVALHO, M.D.G. Alzheimer’s disease: Risk factors and potentially protective measures. J. Biomed. Sci. 2019.
SILVESTRE, C. M. R. F. O diálogo entre o cérebro e o intestino: O papel dos probióticos: Revisão de literatura. Dissertação (Mestrado Integrado em Medicina). – Faculdade de Medicina da Universidade de Lisboa, Lisboa, Portugal, 2016.
SHAW CK, ABDULAI-SAIKU S, MARINO F, WANG D, DAVIS EJ, PANNING B, et al. X chromosome factor Kdm6a enhances cognition independent of its demethylase function in the aging XY male brain. J Gerontol Ser A. 2023:glad007.
SHINJYO N, KITA K. Infection and Immunometabolism in the Central Nervous System: A Possible Mechanistic Link Between Metabolic Imbalance and Dementia. Front Cell Neurosci. 2021 Nov 2;15:765217. Doi: 10.3389/fncel.2021.765217. PMID: 34795562; PMCID: PMC8592913.
SOLLEIRO-VILLAVICENCIO H, RIVAS-ARANCIBIA S (2018). Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front Cell Neurosci, 12:114.
SOUSA T, MOREIRA P, CARDOSO S. Current advances in mitochondrial targeted interventions in Alzheimer’s disease. Biomedicines, 11 (9), 2023.
SUN, Y.-X.; JIANG, X.-J.; LU, B.; GAO, Q.; CHEN, Y.-F.; WU, D.-B.; ZENG, W.-Y.; YANG, L.; LI, H.-H.; Yu, B. Roles of Gut Microbiota in Pathogenesis of Alzheimer’s Disease and Therapeutic Effects of Chinese Medicine. Chin. J. Integr. Med. 2022, 28, 1048–1056
SUN N, YOULE RJ, FINKEL T. The Mitochondrial Basis of Aging. Mol Cell. 2016 Mar 3;61(5):654-666. doi: 10.1016/j.molcel.2016.01.028. PMID: 26942670; PMCID: PMC4779179.
TA TT, DIKMEN HO, SCHILLING S, CHAUSSE B, LEWEN A, HOLLNAGEL JO, KANN O. Priming of microglia with IFN-γ slows neuronal gamma oscillations in situ. Proc Natl Acad Sci U S A. 2019 Mar 5;116(10):4637-4642. Doi: 10.1073/pnas.1813562116. Epub 2019 Feb 19. PMID: 30782788; PMCID: PMC6410786.
TANAKA T, TAKAHASHI K, HIRANO H, KIKUTANI T, WATANABE Y, OHARA Y, et al. Oral frailty as a risk factor for physical frailty and mortality in community-dwelling elderly. J Gerontol A Biol Sci Med Sci. 2018;73(12):1661-7.
TOSTES K, DOS SANTOS AC, ALVES LO, BECHARA LRG, MARASCALCHI R, MACABELLI CH, GREJO MP, FESTUCCIA WT, GOTTLIEB RA, FERREIRA JCB, CHIARATTI MR. Autophagy deficiency abolishes liver mitochondrial DNA segregation. Autophagy. 2022 Oct;18(10):2397-2408. Doi: 10.1080/15548627.2022.2038501. Epub 2022 Feb 27. PMID: 35220898; PMCID: PMC9542960.
TUMURBAATAR B, FRACASSI A, SCADUTO P, GUPTARAK J, WOLTJER R, JUPITER D, TAGLIALATELA G. Preserved autophagy in cognitively intact non-demented individuals with Alzheimer’s neuropathology. Alzheimers Dement. 2023 Dec;19(12):5355-5370. Doi: 10.1002/alz.13074. Epub 2023 May 16. PMID: 37191183; PMCID: PMC10651802.
WANG, L. The emerging mechanisms and functions of microautophagy. Nat Rev Mol Cell Biol 24, 186–203 (2023).
WEAVER DF. Alzheimer’s disease as an innate autoimmune disease (AD2): A new molecular paradigm. Alzheimers Dement. 2023 Mar;19(3):1086-1098. doi: 10.1002/alz.12789. Epub 2022 Sep 27. PMID: 36165334.
WEBERS, A; HENEKA, MT.; GLEESON, PA. The role of innateimmune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunology and Cell Biology. V. 98, n. 1, p. 28–41, 2020.
WEIDLING I.W., SWERDLOW R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020;330:113321.
WEAVER DF. Alzheimer’s disease as na innate autoimmune disease (AD2): A new molecular paradigm. Alzheimers Dement. 2023 Mar;19(3):1086-1098. Doi: 10.1002/alz.12789. Epub 2022 Sep 27. PMID: 36165334.
WEAVER DF. Β-Amyloid is na Immunopeptide and Alzheimer’s is na Autoimmune Disease. Curr Alzheimer Res. 2021;18(11):849-857. Doi: 10.2174/1567205018666211202141650. PMID: 34856900.
WESTFALL, S.; Iqbal, U.; Sebastian, M. & Pasinetti, G.M. Gut microbiota mediated allostasis prevents stress-induced neuroinflammatory risk factors of Alzheimer’s disease. Progress in Molecular Biology and Translational Science. 168:147-181, 2019.
WESTWOOD ML, GEISSMANN Q, O’DONNELL AJ, RAYNER J, SCHNEIDER W, ZUK M, BAILEY NW, REECE SE. Machine learning reveals singing rhythms of male Pacific field crickets are clock controlled. Behav Ecol. 2023 Dec 23;35(1):arad098. Doi: 10.1093/beheco/arad098. PMID: 38144906; PMCID: PMC10748470.
WHILEY L, CHAPPELL KE, D’HONDT E, LEWIS MR, JIMÉNEZ B, SNOWDEN SG, SOININEN H, KŁOSZEWSKA I, MECOCCI P, TSOLAKI M, VELLAS B, SWANN JR, HYE A, LOVESTONE S, LEGIDO-QUIGLEY C, HOLMES E; ADDNEUROMED CONSORTIUM. Metabolic phenotyping reveals a reduction in the bioavailability of serotonin and kynurenine pathway metabolites in both the urine and serum of individuals living with Alzheimer’s disease. Alzheimers Res Ther. 2021 Jan 9;13(1):20. Doi: 10.1186/s13195-020-00741-z. PMID: 33422142; PMCID: PMC7797094.
XIA, Y., XIAO, Y., WANG, ZH. et al. Bacteroides Fragilis in the gut microbiomes of Alzheimer’s disease activates microglia and triggers pathogenesis in neuronal C/EBPβ transgenic mice. Nat Commun 14, 5471 (2023). https://doi.org/10.1038/s41467-023-41283-w
YEAP MW, LOH TC, CHONG MC, YEO WK, GIRARD O, TEE CCL. Influence of Running Velocity and Hypoxic Exposure on Vastus Lateralis Muscle Oxygenation. Int J Sports Physiol Perform. 2024 Sep 4;19(11):1334-1338. Doi: 10.1123/ijspp.2024-0083. PMID: 39231493.
YU L, PETYUK VA, GAITERI C, MOSTAFAVI S, YOUNG-PEARSE T, SHAH RC, BUCHMAN AS, SCHNEIDER JA, PIEHOWSKI PD, SONTAG RL, FILLMORE TL, SHI T, SMITH RD, DE JAGER PL, BENNETT DA. Targeted brain proteomics uncover multiple pathways to Alzheimer’s dementia. Ann Neurol. 2018 Jul;84(1):78-88. doi: 10.1002/ana.25266. Epub 2018 Jul 3. PMID: 29908079; PMCID: PMC6119500.
https://lattes.cnpq.br/1198252075678764