USE OF THE CRISPR-CAS9 TECHNIQUE IN FETAL HEMOGLOBIN REACTIVATION: A POSSIBLE THERAPEUTIC APPROACH FOR THE TREATMENT OF SICKLE CELL ANEMIA THROUGH BCL11A GENE INHIBITION
REGISTRO DOI: 10.69849/revistaft/fa10202511302311
Thaissa Ribeiro Xavier1
Wender Nathan Maas2
Gabriele Marcelino Gomes3
RESUMO
A Anemia Falciforme (AF) é uma doença genética debilitante, caracterizada pela mutação no gene da ẞ-globina (HbS), que leva à falcização das hemácias, anemia hemolítica e crises vasooclusivas. A terapia gênica surge como uma alternativa promissora aos tratamentos tradicionais. Este trabalho consiste em uma revisão narrativa de literatura, com abordagem qualitativa, e objetiva analisar os avanços técnicos e clínicos da aplicação da técnica CRISPRCas9 no tratamento da AF. O foco da análise é a estratégia de reativação da hemoglobina fetal (HbF) por meio da inibição do gene BCL11A. Os estudos analisados demonstram que o BCL11A atua como um repressor da síntese de gamma-globina (componente da HbF) após o nascimento. A edição gênica ex vivo de células-tronco hematopoiéticas (HSPCs) com CRISPR-Cas9 para interromper o intensificador eritroide do BCL11A tem se mostrado eficaz em reativar a produção de HbF. Ensaios clínicos, como o CLIMB-SCD-121, reportam que pacientes tratados apresentaram níveis elevados e sustentados de HbF, resultando na redução significativa ou eliminação de crises vaso-oclusivas. Apesar do potencial curativo, a abordagem enfrenta desafios significativos, incluindo a necessidade de quimioterapia mieloablativa (ex: bussulfano) antes da infusão das células editadas, que possui alta toxicidade, além de eventos adversos observados nos ensaios e questões bioéticas. Conclui-se que a CRISPR-Cas9 é uma ferramenta revolucionária para o tratamento da AF, mas ainda são necessários mais estudos de longo prazo para otimizar a segurança, estabilidade genômica e acessibilidade da técnica antes de sua aplicação clínica generalizada.
Palavras-Chaves: Anemia Falciforme, CRISPR-Cas9, Hemoglobina Fetal, BCL11A, Terapia Gênica.
ABSTRACT
Sickle cell anemia (SCA) is a debilitating genetic disease characterized by a mutation in the βglobin gene (HbS), leading to sickling of red blood cells, hemolytic anemia, and vaso-occlusive crises. Gene therapy emerges as a promising alternative to traditional treatments. This work consists of a narrative literature review, with a qualitative approach, and aims to analyze the technical and clinical advances in the application of the CRISPR-Cas9 technique in the treatment of SCA. The focus of the analysis is the strategy of reactivating fetal hemoglobin (HbF) through the inhibition of the BCL11A gene. The studies analyzed demonstrate that BCL11A acts as a repressor of γ-globin synthesis (a component of HbF) after birth. Ex vivo gene editing of hematopoietic stem cells (HSPCs) with CRISPR-Cas9 to disrupt the erythroid enhancer of BCL11A has proven effective in reactivating HbF production. Clinical trials, such as CLIMB-SCD-121, report that treated patients showed elevated and sustained levels of HbF, resulting in a significant reduction or elimination of vaso-occlusive crises. Despite the curative potential, the approach faces significant challenges, including the need for myeloablative chemotherapy (e.g., busulfan) before the infusion of edited cells, which has high toxicity, as well as adverse events observed in trials and bioethical issues. It is concluded that CRISPRCas9 is a revolutionary tool for the treatment of SCA, but further long-term studies are still needed to optimize the safety, genomic stability, and accessibility of the technique before its widespread clinical application.
Keywords: Sickle Cell Anemia, CRISPR-Cas9, Fetal Hemoglobin, BCL11A, G Therapy
1. INTRODUÇÃO
A terapia genética que inclui a edição e manipulação do genoma humano, a ativação, inibição, reparo ou correção de um lócus do DNA abre a possibilidade de curar doenças genéticas antes consideradas incuráveis. Essa possibilidade tornou-se realidade com a ferramenta CRISPR-Cas9, um mecanismo encontrado em bactérias, como defesa contra elementos invasores (Lins et al., 2018).
Essa técnica permite substituir segmentos da cadeia de DNA por outros, corrigindo erros genéticos ou inserindo características benéficas, seja por inibição ou ativação de determinado códons específicos, conforme a necessidade da intervenção terapêutica (Ramos, 2016; Sganzerla e Pessini, 2020).
Assim, a comunidade científica tem feito estudos acerca da possibilidade, aplicabilidade e eficácia da técnica CRISPR-Cas9 para o tratamento da anemia falciforme, visando curar a doença e melhorar a qualidade de vida dos pacientes, de maneira financeiramente acessível. Isso é relevante, dois frequentemente esses pacientes necessitam submeter-se a tratamentos medicamentosos prolongados e terapias com resultados limitados (Mataveia, 2020).
Entre as doenças hereditárias mais prevalentes no Brasil, destaca-se a anemia falciforme (AF), que atinge cerca 30.000 pessoas, correspondendo a cerca de 8% da população negra (Brasil, 2010). A AF é uma doença hereditária, autossômica recessiva, caracterizada pela mutação no cromossomo 11, que altera o 6º aminoácido da cadeia β-globina e consequente a forma estrutural e química das hemácias, prejudicando o transporte de oxigênio e causando causando crises vaso-oclusivas (Bueno, Da Cruz e Holanda, 2022).
Os avanços em terapias genéticas buscam modular e recapitular o fenótipo da persistência hereditária da hemoglobina fetal. Para isso, utiliza-se a edição genética CRISPRCas9 aplicada em células-tronco e progenitoras hematopoiéticas (HSPCs), visando modificar a região intensificadora eritroide do gene BCL11A (B-cell lymphona/leucemia 11 A) (GUIMARÃE, M, 2016). A redução da expressão de BCL11A permitir restaurar a síntese de γ-globina e reativar a produção de hemoglobina fetal, que possui maior afinidade pelo oxigênio e não sofre as mutações que deformam os eritrócitos (Canver et al., 2015).
A comunidade científica tem utilizado abordagens in vivo e ex vivo para testes clínicos com a CRISPR Cas9. A edição genômica ex vivo consiste na modificação do genoma de células específicas é alterado in vitro, seguida do transporte dessas células modificadas no paciente, visando induzir um efeito regenerativo, (BXet al., 2017). Já na abordagem in vivo, os componentes do sistema CRISPR/Cas9 são diretamente entregues ao ambiente celular para realizar seus efeitos benéficosin situ. (Bioconjugate Chem, 2017)
Apesar dos avanços biotecnológicos, a edição gênica enfrenta obstáculos relativos à bioética, principalmente quanto ao limite da interferência científica nos genes, tanto in vivo quanto ex vivo (Nascimento, 2020). Ademais, a administração do CTX001, utilizada para aumentar a hemoglobina fetal e inibir o gene BCL11A, tem apresentado eventos adversos durante os ensaios clínicos no tratamento da anemia falciforme, o que tem ocasionado atrasos no desenvolvimento da terapia. Entre os efeitos colaterais relatados destacam-se pneumonia associada à neutropenia, síndrome de obstrução venosa hepática (VOD-SOS), sepse associada à neutropenia, colelitíase e dor abdominal. Também foram observados eventos adversos não graves, como linfopenia (Bomberger, 2022).
Os ensaios clínicos demonstraram que a utilização da edição gênica CRISPR-Cas9 pode revolucionar o tratamento da anemia falciforme, com resultados positivos (Frangoul et al., 2021).
A técnica CRISPR-Cas9 para o tratamento da anemia falciforme é uma abordagem promissora, mas ainda está em fase de ensaios clínicos. Até o momento, não está disponível para o público geral, e seu uso é restrito a participantes de estudos clínicos. A expectativa é que, com a conclusão dos ensaios e a aprovação regulatória, essa terapia possa se tornar uma opção viável para pacientes em breve.
O presente estudo não necessitou de submissão ao Comitê de Ética em Pesquisa, uma vez que se trata de uma revisão bibliográfica. Não houve envolvimento direto de seres humanos ou animais, tampouco coleta de dados primários, limitando-se à análise e discussão de informações já publicadas na literatura científica.
2. REFERENCIAL TEÓRICO
2.1 Estrutura das Hemácias
A hemoglobina é uma proteína de ligação ao oxigênio encontrada nos eritrócitos que transporta oxigênio dos pulmões para os tecidos. A síntese para a cadeia alfa está no cromossomo 16, e os genes para a cadeia beta estão no cromossomo 11. Cada molécula de hemoglobina é um tetrâmero composto por quatro cadeias polipeptídicas de globina. Cada subunidade de globina contém uma porção heme formada por um anel orgânico de protoporfirina e um íon de ferro central no estado ferroso (Fe₂+) como ilustra na (Figura 1). A molécula de ferro em cada porção heme pode ligar e desligar o oxigênio, permitindo o transporte de oxigênio no corpo (Hafen, 2022).
Figura 1- Estrutura tetrâmera da hemoglobina
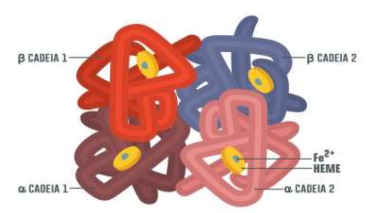
Fonte: Adaptado de CUREM (2021). Nota: O grupo heme – formado por Protoporfirina IX + Ferro – é o grande responsável pela ligação com o oxigênio. Sendo assim, qualquer condição que leve à produção anormal de alguma dessas cadeias pode alterar não apenas a forma da hemácia, mas também a afinidade da Hb ao oxigênio.
Existem algumas formas diferentes de hemoglobina normal no sangue humano. A prevalência percentual de cada tipo de hemoglobina depende do estágio de desenvolvimento (Hafen, 2022). No entanto alguns fatores genéticos podem influenciar diretamente na produção da HbA e de suas cadeias de globina, alterando dessa forma síntese e causando polimorfismo nas células eritrocitárias, diminuindo de forma significativa sua função metabólica, que ocorre gradualmente na anemia falciforme. (Chiabrando, Mercúrio, Tolosano, 2014).
2.2 Fisiopatologia da Anemia Falciforme
A anemia falciforme (AF) é uma condição hereditária caracterizada pela presença de um defeito estrutural na molécula de hemoglobina, resultando em uma anemia hemolítica. A raiz da doença é uma mutação pontual no gene da beta globina, que leva à produção de uma forma anormal de hemoglobina, denominada hemoglobina S (HbS), ao invés da hemoglobina A (HbA) considerada saudável (Ladeia, 2020).
Essa alteração genética resulta na troca do ácido glutâmico pela valina na sexta posição da sequência beta do gene, causando transformações nas propriedades físico-químicas da estrutura da hemoglobina (Figura 2). Indivíduos afetados pela anemia falciforme são homozigotos para essa mutação, exibindo o perfil eletroforético compatível com a hemoglobina SS (HbSS) (Ladeia, 2020).
Figura 2- Estrutura da hemoglobina e a falciformação das hemácias
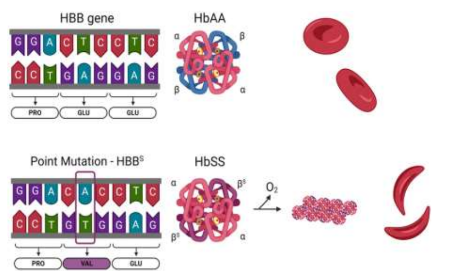
Fonte: Duyuran, R.; Kanbur Çiçek, H, (2024). Notas: Substituição de GAG para GTG no DNA, resultando na troca do aminoácido glutamato por valina na hemoglobina.
Porém, quando há alteração na forma estrutural das hemoglobinas, a variante desoxigenada da hemoglobina S (HbS) desencadeia a polimerização hemoglobínica, levando à perda da organização quaternária e à adoção de uma organização primária. Esse processo insolubiliza a HbS, causando uma transformação nas hemácias saudáveis em uma forma semelhante a uma foice, conhecida como eritro falciformação, ou hemácias em forma de drepanócitos (Figura 3). Isso resulta em obstruções microvasculares generalizadas, causando danos isquêmicos aos tecidos e crises de dor, chamadas crises vaso-oclusivas. Além disso, ocorre anemia crônica devido à hemólise intravascular e extravascular (Zago, 2020).
Figura 3- Hemácias em forma de drepanócitos
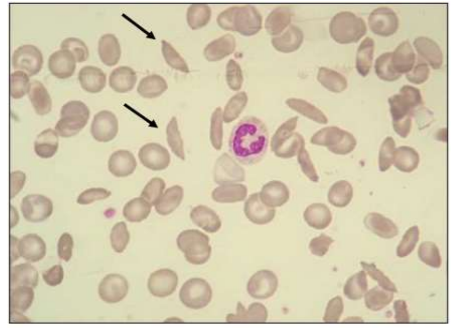
Fonte: ROMANA, Danyelle (2020). Nota: Em um esfregaço sanguíneo nota-se a presença de poiquilocitose nas hemácias, algumas delas em forma de drepanócitos, característica própria da anemia falciforme.
Quando expostas à baixa tensão de oxigênio, a polimerização acontece e, por sua vez, causa desidratação mediada pelo influxo de cálcio, elevando a concentração de hemoglobinas intraeritrocitárias. Assim, a célula torna-se mais rígida e alongada e o sangue mais viscoso, fatores que impedem a livre passagem do fluxo pelos vasos sanguíneos mais estreitos (Figura 4). Como resultado, ocorre a vaso-oclusão e, consequentemente, infarto tecidual, além de estimular uma resposta inflamatória (Steinberg, 2008).
Figura 4- Fisiopatologia da Anemia Falciforme
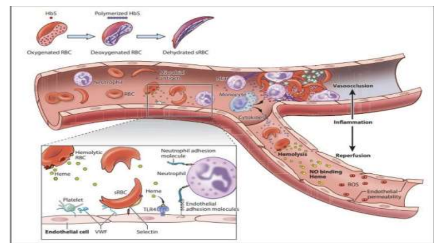
Na anemia falciforme, a polimerização da hemoglobina S (HbS) pode levar à produção de espécies reativas de oxigênio (EROs), que, em conjunto com o aumento dos níveis de cálcio intracelular, contribuem para a exposição da fosfatidilserina (PS) na membrana das hemácias. A PS é um aminofosfolipídio que, em condições fisiológicas, encontra-se voltada para o citoplasma, mantendo-se na face interna da membrana por ação da enzima flipase (Keohane, 2016).
Essa organização garante a assimetria da bicamada lipídica. No entanto, os repetidos ciclos de falcização e desfalcização das hemácias falciformes causam alterações estruturais na membrana, provocando a redistribuição dos fosfolipídios e exteriorizando a PS (Stypulkowski; Manfredini, 2010).
A presença da PS na superfície externa das hemácias é reconhecida por macrófagos do sistema mononuclear fagocitário, levando à sua fagocitose precoce e à intensificação do quadro hemolítico. Além disso, a PS ativada pode desencadear processos de coagulação, estimular plaquetas, aumentar a expressão de moléculas de adesão no endotélio vascular e favorecer a obstrução dos vasos sanguíneos. Também se torna alvo da ação da fosfolipase A2 (PLA2), que produz ácidos graxos pró-inflamatórios, colaborando com danos vasculares e com o desenvolvimento da Síndrome Torácica Aguda (STA), uma complicação grave da doença falciforme (Keohane, 2016; Stypulkowski; Manfredini, 2010).
2.2.3 Sinais e sintomas da AF
A partir do sexto mês de vida, observa-se uma queda nos níveis de hemoglobina fetal (HbF) e um aumento progressivo da hemoglobina S (HbS), o que marca o início dos primeiros sinais clínicos da doença falciforme. Nesse contexto, as hemácias tornam-se suscetíveis à hemólise intensa, desencadeando anemia hemolítica e esplenomegalia, geralmente associada ao sequestro esplênico (Keohane, 2016).
A crise de sequestro esplênico compromete a função esplênica de forma progressiva, levando à ocorrência de hipoesplenismo ou até mesmo asplenismo em adultos (Lorenzi, 2003). A vaso-oclusão, por sua vez, é a principal causa de dor intensa em articulações e ossos, sendo um dos principais motivos de internações hospitalares. Essas crises podem surgir espontaneamente, mas são frequentemente desencadeadas por fatores como infecções, febre, desidratação e exposição ao frio (LOBO, 2007).
Entre as complicações crônicas mais graves está o acidente vascular cerebral (AVC), que atinge cerca de 25% dos adultos com a doença. Esse evento está relacionado à estenose e oclusão vascular, podendo causar prejuízos cognitivos mesmo na ausência de sinais neurológicos clínicos evidentes (Neumayr et al., 2019; STEINBERG, 2008). Outra complicação de destaque é a Síndrome Torácica Aguda (STA), que pode acometer pacientes de todas as idades e se manifesta por febre, dor torácica, taquipneia, tosse e desconforto generalizado, estando associada a infecções, embolias e podendo ser fatal (LORENZI, 2003).
2.3 Abordagens terapêuticas atuais
A descoberta de novos compostos capazes de prevenir essa polimerização tem sido um foco relevante na pesquisa atual. O voxelotor é um medicamento recentemente aprovado que atua estabilizando a hemoglobina, impedindo a formação de hemácias falciformes. Outro tratamento promissor é o crizanlizumabe, um anticorpo monoclonal que se liga à P-selectina, proteína envolvida nas crises vaso-oclusivas, reduzindo significativamente os episódios dolorosos em pacientes com anemia falciforme (BELL et. al, 2024).
Além dessas terapias farmacológicas, o transplante alogênico de células-tronco hematopoiéticas, também conhecido como transplante de medula óssea, permanece como o único tratamento curativo comprovado para a doença. Realizado preferencialmente com doadores HLA-compatíveis, como irmãos não afetados, esse procedimento tem apresentado melhores taxas de sucesso devido aos avanços nas técnicas de transplante e na ampliação da compatibilidade entre doadores (BELL et al., 2024).
O transplante de células-tronco hematopoiéticas (TCTH), é atualmente a técnica comprovada cientificamente que pode trazer a cura a anemia falciforme, mas sofre alguns obstáculos no que se refere a doadores compatíveis de medula óssea. Logo, um doador saudável sem variante patogênica HBB pode ser curativo em indivíduos com anemia falciforme. Crianças com anemia falciforme que recebem TCTH usando um doador irmão compatível podem esperar mais de 90% de chance de cura com uma sobrevida global de 93% (Gluckman et al., 2017).
No Quadro 1 podemos analisar as correlações das terapias utilizadas atualmente na doença falciforme, pontuando os benefícios e suas objeções ao longo dos testes, além de destacar a técnica de edição gênica como uma ferramenta promissora na cura da AF.
Quadro 1- Terapias atuais para AF
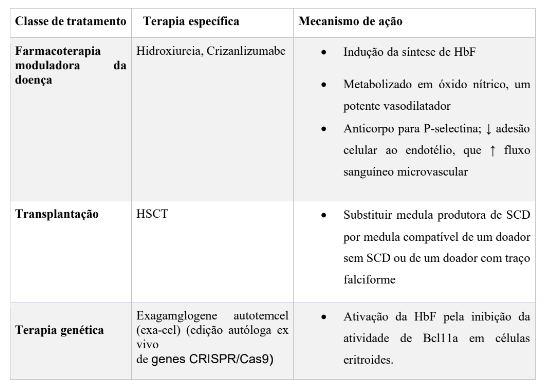
2.4 Terapia gênica CRISPR Cas9
2.4.1 Mecanismo geral da técnica CRISPR Cas9
O sistema de nuclease Cas9 (CRISPR)- repetições palindrômicas curtas regularmente espaçadas em cluster, um sistema imunológico bacteriano que pode clivar DNA de bacteriófagos ou plasmídeos, permite o direcionamento programável de inserções ou exclusões (indels) em um local específico do DNA genômico (Bak RO, Dever DP, 2018).
Essa técnica utiliza apenas três moléculas: a nuclease (Cas9), que corta a dupla fita de DNA; um RNA guia que direciona o complexo até o alvo desejado; e o próprio DNA alvo. Comparada a outras técnicas, como Zinc-Finger Nucleases, TALENs e Gene Targeting, o sistema CRISPR se destaca por sua simplicidade e precisão (Figura 5). Isso faz com que ele seja uma ferramenta versátil para diferentes abordagens de edição gênica, incluindo inativação gênica (gene knockout-KO), inserção de sequências exógenas e substituição alélica, entre outras (HÖIJER, 2022).
Figura 5- Mecanismo geral da CRISPR Cas9
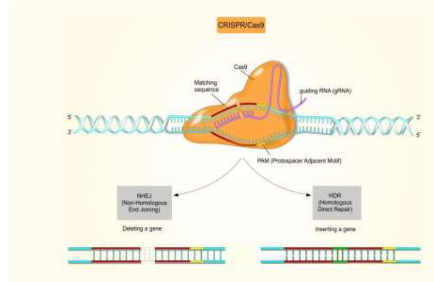
Fonte: ZARA, puckrin (2021). Nota: Codificações simples por knock-out podem ser realizadas com muito mais facilidade usando o CRISPR Cas9, é mais intuitivo do que as tecnologias de edição genômica disponíveis anteriormente.
A ferramenta CRISPR Cas9 tem se consolidado como uma das principais tecnologias de edição genética, permitindo aplicações in vivo e ex vivo. No entanto, as abordagens in vivo ainda enfrentam limitações, sobretudo quanto aos métodos de entrega do sistema, o que tem motivado o estudo de diferentes estratégias para viabilizar sua aplicação segura e eficaz (LUTHER et al., 2018).
Os vetores virais continuam sendo os mais utilizados para a entrega do sistema CRISPR Cas9, destacando-se os lentivíruses, adenovírus (AV) e adenovírus associados (AAV). Apesar da alta eficiência, muitos desses vetores podem desencadear resposta imune no organismo hospedeiro. Os AAVs, por sua vez, têm demonstrado melhor perfil de segurança, sendo considerados mais apropriados para terapias gênicas in vivo (LUTHER et al., 2018). A entrega via RNA mensageiro (RNAm) possui como vantagem a não integração permanente ao genoma e uma rápida expressão do sistema, mas requer a administração separada de cada componente e enfrenta limitações devido à instabilidade do RNA, o que pode comprometer sua eficácia (LUTHER et al., 2018).
Outra abordagem é o uso direto do complexo ribonucleoproteico Cas9-RNP, formado pela nuclease Cas9 e o RNA guia. Essa técnica tem demonstrado alta especificidade, baixa taxa de mutações fora do alvo e reduzida resposta imune, embora ainda enfrente barreiras celulares, como a degradação endossomal e o curto tempo de atividade intracelular (LUTHER et al., 2018).
2.5 Edição gênica na anemia falciforme
2.5.1 Hemoglobina fetal HbF e sua correlação no tratamento da anemia falciforme.
Estudos de associação genômica identificaram polimorfismos de nucleotídeo único (SNPs) no locus BCL11A, localizado no cromossomo 2, que estão correlacionados com o aumento da expressão da hemoglobina fetal (HbF) em adultos. Esses SNPs afetam a atividade do gene BCL11A, que codifica um fator de transcrição com domínios de dedo de zinco. O BCL11A atua como um repressor transcricional, inibindo a expressão dos genes da γ-globina e, consequentemente, suprimindo a produção de HbF em células eritroides (SANKARAN et al., 2011; MANKU et al., 2021; (BAUER et al., 2013; CANVER et al., 2015).
Os SNPs associados à HbF estão localizados em um intensificador específico de eritroides, que regula negativamente a expressão do BCL11A. Essa modulação permite uma maior expressão dos genes da γ-globina, promovendo o aumento da síntese de hemoglobina fetal. Essa manutenção de níveis elevados de HbF em adultos tem sido associada à redução da gravidade clínica de doenças hemolíticas, como a doença falciforme. O conhecimento sobre os mecanismos genéticos que regulam a expressão da HbF abre perspectivas para o desenvolvimento de terapias direcionadas, visando modular a atividade de fatores como o BCL11A (Steinberg MH, 2021; Steinberg MH, 2020).
2.6 Edição gênica aplicada na anemia falciforme
Recentemente, ensaios clínicos têm explorado a utilização de CRISPR para editar células-tronco hematopoiéticas dos pacientes, corrigindo a mutação causadora da AF antes de reimplantá-las nos pacientes. Esta abordagem mostrou potencial significativo para uma cura permanente (BELL et al., 2024)
Em uma tentativa de recapitular o fenótipo da persistência hereditária da hemoglobina fetal, técnicas de edição genética CRISPR-Cas9 em células-tronco e progenitoras hematopoiéticas (HSPCs) são utilizadas na região intensificadora específica de eritroides de BCL11A para reduzir a expressão de BCL11A em células da linhagem eritróide, desta forma restaurando a síntese de γ-globina e reativando a produção de hemoglobina fetal (Canver MC, 2019).
2.7 Edição com uso de CRISPR Cas9
Para a edição dos promotores de hemoglobinas no tratamento da anemia falciforme, SHARMA (2023) cita que a triagem de CRISPR-Cas9 em mosaico. Estes promotores foram produzidos por eletroporação de células CD34+. Estas foram obtidas de doadores saudáveis e avaliados a fração dos eritroblastos imunomarcados pela hemoglobina fetal.
2.8 A Técnica de CRISPR Cas9 no tratamento da Anemia Falciforme
Segundo o autor Abraham AA (2021), descreve que as células troncos hematopoiéticas (chamadas de HSCs), são coletadas por um procedimento em que o sangue do doador é retirado por um equipamento automatizado para separação dos hemocomponentes (aférese). Essas células então, são editadas por um sistema de CRISPR e então direcionadas para romper uma região celular que foi determinada como intensificadora eritroide de BCL11A. Esta, levou a indução de HbF (Figura 9).
Figura 09: Terapia genética para anemia falciforme: do laboratório para o leito do paciente.
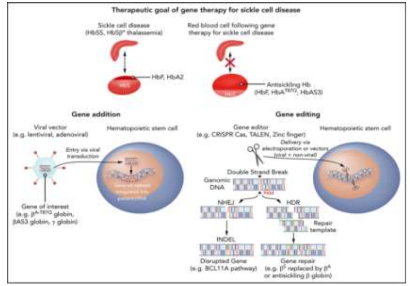
Fonte: Abraham AA (2021). Nota: Comparação entre uma adição gênica para tratar a anemia falciforme.
Em outra, como corrigir um gene defeituoso ou reativar um gene protetor (como o da hemoglobina fetal). Essas duas técnicas, são de adição e de edição dos genes. A adição de genes envolve a inserção de um gene de interesse (como um gene de globina) em uma célula-tronco hematopoiética usando um vetor viral. A edição de genes utiliza editores de genes como CRISPR-Cas para criar uma quebra de fita dupla no DNA genômico. A edição pode resultar em um gene interrompido (via NHEJ) ou em um reparo de gene (via HDR), que permite a substituição do gene defeituoso. Ambas as abordagens visam corrigir o defeito genético subjacente que causa a doença falciforme, resultando na produção de hemoglobina “antifalciforme”. Estas células geneticamente modificadas, são então infundidas após a mieloblação com bussulfano (Figura 10).
Figura 10: Terapia genética para anemia falciforme: do laboratório para o leito do paciente.
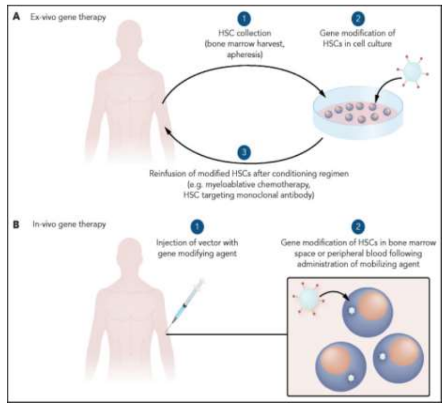
Fonte: Abraham AA (2021). Nota: explanação de uma terapia ex vivo e in vivo. A primeira seria a correção com a remoção das células do paciente, correção em laboratório, e devolução ao corpo. A segunda, seria a correção pela injeção do tratamento no corpo do paciente.
A terapia ex-vivo envolve a modificação genética fora do paciente. As células-tronco hematopoiéticas (HSCs) são coletadas, modificadas em laboratório e então reinfundidas no paciente. Já a terapia in-vivo ocorre diretamente no corpo do paciente. Nesse caso, o agente modificador é administrado no paciente, localizando e realizando a modificação genética nas células-alvo dentro do organismo. Um agente mobilizador pode ser usado para fazer as HSCs saírem da medula óssea e ficarem mais acessíveis ao vetor na terapia in-vivo. A principal diferença entre as duas abordagens é o local onde a modificação genética é realizada: ex-vivo é feita em laboratório, enquanto in-vivo acontece diretamente no corpo do paciente após a administração do agente (ABRAHAM, 2021).
3 METODOLOGIA
A pesquisa relacionada neste estudo, se caracteriza por uma revisão literária, de modo qualitativo. Seu principal objetivo é analisar os avanços técnicos científicos e clínicos. Estes, relacionados à aplicação da técnica de edição gênica CRISPR-Cas9 no tratamento da anemia falciforme.
A abordagem, se optou por uma revisão narrativa. Visando descrever, reunir e discutir os estudos principais disponíveis na literatura nacional, quanto na literatura internacional. Uma revisão narrativa, nada mais é do que publicação ampla, usada para descrever e discutir o desenvolvimento de algum determinado assunto. Esse pode ser usado um ponto de vista teórico ou contextual (ROTHER, 2007).
Já para a coleta de dados, foram usadas buscas sistemáticas e sites dedicados a publicações científicas. Entra essas, estão PubMed, Scopus, Web of Science, Embase, Medline e SciELO, utilizando os seguintes descritores combinados por operadores booleanos: “CRISPR-Cas9” AND “Sickle Cell Disease”, “Gene Therapy” AND “Hemoglobinopathies”, “BCL11A” AND “HbF induction”, entre outros termos equivalentes em português e inglês.
Os critérios de inclusão, foram os artigos publicados nos últimos 5 anos, estudos originais, ensaios clínicos e artigos de revisão com relevância comprovada. Também foram usados trabalhos que abordam diretamente a aplicação da CRISPR-Cas9 em modelos in vivo ou ex vivo para a correção da mutação da anemia falciforme ou ainda, a indução da hemoglobina fetal (HbF). Também foi levado em consideração artigos integralmente em português ou em inglês que sejam gratuitos.
Como critérios de exclusão, usamos artigos que estão duplicados na base de dados coletada, trabalhos com foco em outras doenças, resumos de congressos se, publicação completa, ou ainda, estudo sem rigor metodológico evidente.
Ainda em nossa análise, revisamos cerca de 20 artigos para embasamento teórico. Os critérios formam relevância e ainda, artigos publicados recentemente.
Esta análise de dados foi conduzida de forma qualitativa com uma categorização temática das abordagens identificadas como: ativa, com categorização temática das abordagens terapêuticas identificadas, como: (1) modulação do gene BCL11A; (2) reativação da HbF via edição dos promotores de HBG1/HBG2; (3) correção direta da mutação no gene HBB; (4) uso de tecnologias alternativas como prime editing e base editing.
4. RESULTADOS E DISCUSSÃO
Os resultados apresentados nos estudos analisados evidenciam avanços significativos na aplicação da técnica CRISPR-Cas9 para o tratamento da anemia falciforme, especialmente quanto à modulação do gene BCL11A e à reativação da hemoglobina fetal (HbF).
No estudo, três participantes receberam o produto de células-tronco hematopoiéticas (OTQ923) autólogo após o condicionamento mieloblativo. Ao serem acompanhados por até 18 meses, os participantes tiveram uma indução da hemoglobina fetal entre 19% e 26,8%.
Ainda, as taxas de anemia falciforme diminuiram durante o acompanhamento (SHARMA, Akshay et. al, 2023).
Ainda seguindo nesta linha, Abraham AA (2021) descreve um ensaio. Neste Ensaio chamado de CLIMB-SCD-121, as células tronco hematopoiéticas (chamadas de HSCs), são coletadas por um procedimento em que o sangue do doador é retirado por um equipamento automatizado para separação dos hemocomponentes (aférese). Essas células então, são editadas por um sistema de CRISPR. Foram então direcionadas para romper uma região celular que foi determinada como intensificadora eritroide de BCL11A. Esta, levou a indução de HbF. Estas células geneticamente modificadas, são então infundidas após a mieloblação com bussulfano. O estudo de Abraham et al. (2021) relata que, 15 meses após o tratamento, o paciente estava livre de eventos que caracterizavam a doença vaso-oclusiva. Além disso, observou-se um aumento significativo nos níveis de hemoglobina fetal (HbF) para 43,1%, em comparação com os níveis anteriores de 9,1%. Houve também uma redução no nível de hemoglobina S (HbS) de 74,1% para 52,3%, e a hemoglobina total aumentou de 7,1 g/dL para 12 g/dL.
Essa estratégia mostrou ser capaz de reduzir expressivamente os sintomas clínicos e as crises vaso-oclusivas nos pacientes testados. No entanto, observa-se que muitos desses resultados ainda estão restritos a ensaios clínicos iniciais e estudos experimentais, o que limita a extrapolação dos dados para a prática clínica em larga escala. A consistência dos achados reforça o potencial terapêutico, mas também evidencia que a consolidação dessa abordagem depende de análises mais amplas e de longo prazo.
Em termos de eficácia, os estudos indicam que a reativação da HbF por meio da edição gênica oferece resultados promissores, pois permite a substituição funcional da hemoglobina anormal (HbS) por uma forma não falciforme. Entretanto, ainda há variabilidade na resposta entre os pacientes, o que sugere que fatores genéticos individuais, condições clínicas preexistentes e o próprio processo de modificação celular podem interferir na estabilidade e na durabilidade da resposta terapêutica. Esses elementos reforçam a necessidade de protocolos personalizados e de maior controle sobre os mecanismos de reparo do DNA após a edição, para evitar mutações fora do alvo.
Os dados clínicos revisados, especialmente os referentes aos ensaios CLIMB-SCD-121 e CLIMB-THAL-111, mostraram resultados consistentes em termos de redução de crises vasooclusivas e aumento significativo de HbF. Contudo, a ocorrência de eventos adversos, como neutropenia, dor abdominal e complicações hepáticas, indica que a técnica ainda necessita de ajustes para minimizar riscos e pacientes que receberam uma única infusão venosa de CTX001. Os pacientes foram limitados a uma eligibilidade de 18 a 35 anos (Frangoul, Et al., 2021).
No ensaio CLIMB THAL-111, os pacientes que participaram que tivessem recebido o diagnóstico de β-talassemia com mutações homozigóticas ou heterozigóticas compostas, teriam que ter recebido compostos de hemácias por transfusão de pelo menos 100 ml por kg de peso corporal por ano durantes os 2 anos anteriores.
Já no ensaio CLIMB SCD-121, estes pacientes que participaram teriam que ter o genótipo βS/βS ou βS/β 0 documentado. Ou ainda, que tivessem o histórico de dois ou mais que dois vaso-oclusivos graves por ano, nos 2 anos anteriores (Frangoul, Et al., 2021).
Já para a produção e infusão, foram coletadas dos pacientes células-tronco e progenitoras hematopoiéticas CD34+. Estas, encontram-se no sangue periférico mobilizado, medula óssea e no sangue do cordão umbilical. Neste caso, foram coletadas por aférese com mobilização de filgrastim e plerixafor.
Estes 2, atuam ligando em receptores nas células progenitoras na medula óssea. Assim, estimulam sua proliferação, se diferenciando e em um aumento significativo na corrente sanguínea (BRASIL, 2021). Estes foram administrados após um mínimo de 8 semanas de transfusões de concentrados de hemácias. Isso, para visar um nível de hemoglobina falciforme de mesmo de 30% (Frangoul, Et al., 2021).
O CTX001 foi feito a partir de células CD34+ por edição de CRISPR-Cas9. Onde foi usado o sequenciamento de DNA para avaliar a porcentagem de edição no sítio alvo. Assim sendo, os pacientes então receberam mieloblação com bulssulfano antes da infusão de CTX001. Ainda houve o monitoramento dos pacientes enquanto a presença do enxerto, e eventos adversos. Dessa maneira, os aspirados da medula óssea foram obtidos de 6 e 12 meses após essa infusão. O sequenciamento de DNA foi usado para medir a fração total de DNA editada no sítio alvo na medula óssea e no sangue periférico (Frangoul, Et al., 2021).
Os resultados pré-clínicos de edição de BCL11A, apontaram a avaliação da frequência de edição genética de células-tronco e progenitoras hematopoiéticas. Uma média de 80% foi observada e mantida em todas as subpopulações de células hematopoiéticas de longo prazo (Frangoul, Et al., 2021).
Outra terapia que pode ser utilizada é a terapia celular chamada de exagamglogene autotemcel (exa-cel). Esta foi desenvolvida para então reativar a síntese da hemoglobina fetal por meio de uma edição ex vivo de repetições palindrômicas curtas e regularmente espaçadas, a CRISPR-Cas9 (FRANGOUL, Haydar, Et al., 2024). O estudo então citado, foi de fase 3 de grupo único com paciente de 12 a 35 anos de idade com anemia falciforme. Estes, apresentaram pelo menos duas crises vaso-oclusivas graves em cada um dos 2 anos anteriores à triagem. As células HSPCs CD34+ foram editados com o uso de CRISPR-Cas9. Estes pacientes foram também submetidos para com o condicionamento mieloblativo com bussulfano com a dose ajustada para cada paciente.
De acordo com FRANGOUL (2024), o desfecho que se suscedeu, foi nos primeiros 12 meses consecutivos a ausência de crises vaso-oclusivas graves. Ainda, houve um desfecho de hospitalização nestes 12 meses.
5. CONSIDERAÇÕES FINAIS
A análise dos estudos selecionados demonstra que a edição gênica por meio da técnica CRISPR-Cas9 representa um avanço promissor para o tratamento da anemia falciforme, especialmente pela sua capacidade de modular o gene BCL11A e reativar a síntese da hemoglobina fetal (HbF). A restauração dessa forma de hemoglobina reduz os efeitos da mutação da β-globina, melhora o transporte de oxigênio e atenua as crises vaso-oclusivas, proporcionando melhora clínica significativa em modelos experimentais e nos ensaios clínicos em andamento.
Os resultados obtidos até o momento evidenciam que a modificação de células-tronco hematopoiéticas ex vivo utilizando o sistema CRISPR-Cas9 permite uma reexpressão sustentada de HbF sem alterações genéticas indesejadas significativas. Contudo, os dados ainda são limitados, e a aplicação clínica ampla requer cautela quanto à segurança, à estabilidade genômica e aos possíveis efeitos adversos, como reações inflamatórias e complicações hematológicas observadas em alguns pacientes tratados.
Os estudos analisados reforçam que a CRISPR-Cas9 pode redefinir as abordagens terapêuticas para doenças monogênicas, oferecendo uma alternativa potencialmente curativa, mas que ainda depende de pesquisas aprofundadas, testes clínicos de longo prazo e investimentos contínuos. O uso dessa biotecnologia deve respeitar os princípios da bioética, a legislação vigente e o controle rigoroso dos protocolos clínicos para garantir a segurança dos indivíduos e a integridade científica das aplicações terapêuticas.
Em síntese, os resultados encontrados demonstram que a CRISPR-Cas9 é uma das ferramentas mais revolucionárias da biomedicina contemporânea, com alto potencial para modificar o tratamento de doenças genéticas como a anemia falciforme. No entanto, o estado atual das pesquisas revela que a técnica ainda não atingiu maturidade clínica suficiente para uma aplicação ampla e segura. As evidências indicam progresso, mas também alertam para a necessidade de monitoramento contínuo, aperfeiçoamento metodológico e fortalecimento da regulação ética e científica antes que a edição gênica possa ser considerada uma alternativa terapêutica consolidada e acessível à população, mas indicam que a consolidação dessa técnica na prática clínica ainda exige a superação de desafios técnicos e éticos antes de ser aplicada de forma segura e acessível à população.
6. REFERÊNCIAS BIBLIOGRÁFICAS
ABRAHAM, A. A.; TISDALE, J. F. Gene therapy for sickle cell disease: moving from the bench to the bedside. Blood, v. 138, n. 11, p. 932-941, 16 set. 2021. DOI: https://doi.org/10.1182/blood.2019003776. Disponível em: https://pubmed.ncbi.nlm.nih.gov/34232993/. Acesso em: 10 set. 2025.
BRASIL, Ministério da Saúde. Doença falciforme: conhecer para cuidar. Secretaria de Vigilância em Saúde, Coordenação Geral de Sangue e Hemoderivados, DAET, SAS, Universidade Federal de Santa Catarina, 2015. Disponível em: https://telelab.aids.gov.br/moodle/ pluginfile.php/39506/mod_resource/content/4/Doe n ca%20Falciforme_SEM.pdf/. Acesso em: 25 de agosto de 2025.
BRASIL. Ministério da Saúde. Agência Nacional de Vigilância Sanitária. FIL (filgrastim) – Novo medicamento. Publicado em 20 de maio de 2022. Disponível em: https://www.gov.br/anvisa/pt-br/assuntos/medicamentos/novos-medicamentos-eindicacoes/fil-filgrastim-novo-medicamento. Acesso em: 7 set. 2025.
BENDER MA, CARLBERG K. SICKLE CELL DISEASE.]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1377/;. 2003.
CANGUVEN, O. et al. Derivação corporoglanular peniana distal transitória como adjuvante aos procedimentos de aspiração e irrigação no tratamento do priapismo isquêmico precoce. Korean Journal of Urology, v. 54, p. 394-398, 2013. [Artigo gratuito do PMC] [PubMed]. Acesso em: 07 de agosto de 2025.
EDEL, Y.; MAMET, R. Porfiria: O que é e quem deve ser avaliado? Rambam Maimonides Medical Journal, 19 de abril de 2018; 9(2). [Artigo gratuito do PMC] [PubMed]. Acesso em: 02 de agosto de 2025.
FRANGOUL, H. et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and βThalassemia. The New England Journal of Medicine, v. 384, p. 252-260, 2021. Disponível em:https://www.nejm.org/doi/full/10.1056/NEJMoa2031054. Acesso em: 18 de agosto de 2025.
GUIMARÃES, M. Uma ferramenta para editar o DNA. Sistema copiado de bactérias, CRISPR-Cas9 pode catalisar descobertas em biologia e medicina e suscita temores éticos. Pesquisa Fapesp, 2016. Disponível em: https://revistapesquisa.fapesp.br/2016/02/19/uma-ferramenta-para-editar-o-dna/. Acesso em: 10 de junho de 2025.
HERRICK, James B. Peculiar Elongated and Sickle-shaped Red Blood Corpuscles in a Case of Severe Anemia. Yale Journal of Biology and Medicine, v. 74, p. 179–184, 2001. Disponível em:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2588723/. Acesso em: 25 de agosto de 2025.
KATO, G. J. Anemias falciformes e traço falciforme na trombose. Sangue, v. 133, p. 2463, 2019. [Artigo gratuito do PMC] [PubMed]. Acesso em: 07 de setembro de 2025.
KEOHANE, E. M.; SMITH, L. J.; WALENGA, J. M. Rodak’s Hematology: Clinical Principles and Applications. 5. ed. St. Louis (Missouri – EUA), 2016. Acesso em: 22 de junho de 2025.
LOBO, Clarisse; MARRA, Vera N.; SILVA, Regina M. G. Crises dolorosas na doença falciforme. Revista Brasileira de Hematologia e Hemoterapia, v. 29, n. 3, p. 247-258, 2007. Disponível em:https://doi.org/10.1590/S1516-84842007000300011. Acesso em: 3 de setembro de 2025.
MATTHEWS, Kerryn et al. Technologies for measuring red blood cell deformability. Lab Chip, v. 22, p. 1254-1274, 2022. Disponível em: https://doi.org/10.1039/d1lc01058a. Acesso em: 20 de junho de 2023.
MA Bender, MD, PhD e Katie Carlberg, MD.Anemia falciforme. Universidade de Washington, Seattle. Gene Reviews 1993-2025. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK1377/. Acesso em: 05 de outubro de 2025.
NATIONAL ACADEMY OF SCIENCES; NATIONAL ACADEMY OF MEDICINE. Human Genome Editing: Science, Ethics and Governance Washington. Washington: National Academies Press, 2017. Acesso em: 04 de junho de 2025.
NEUMAYR, Lynne D.; HOPPE, Carolyn C.; BROWN, Clark. Sickle Cell Disease: Current Treatment and Emerging Therapies. The American Journal of Managed Care, v. 25, n. 18, s. 335-343, 2019. Disponível em:https://www.ajmc.com/view/sickle-cell-disease-currenttreatment-and-emergingtherapies. Acesso em: 10 de agosto 2023.
POLACK, Fernando P. et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. The New England Journal of Medicine, v. 383, n. 27, p. 2603-2615, 31 dez. 2020. DOI: 10.1056/NEJMoa2031054. Disponível em: https://www.nejm.org/doi/full/10.1056/NEJMoa2031054. Acesso em: 7 set. 2025. STEINBERG, Martin H. Sickle Cell Anemia, the First Molecular Disease: Overview of Molecular Etiology, Pathophysiology, and Therapeutic Approaches. The Scientific World Journal, v. 8, p. 1295–1324, 2008. Disponível em: https://doi.org/10.1100/tsw.2008.157. Acesso em: 18 de agosto de 2025.
SGANZERLA, Anor; PESSINI, Leo. Edição de humanos por meio da técnica do Crisprcas9: entusiasmo científico e inquietações éticas. Saúde em Debate, v. 44, p. 527-540, 2020. Disponível em: https://www.scielosp.org/article/sdeb/2020.v44n125/527-540/pt/. Acesso em: 25 de set. 2025.
SHARMA, Akshay et al. CRISPR-Cas9 Editing of the HBG1 and HBG2 Promoters to Treat Sickle Cell Disease. The New England Journal of Medicine, v. 389, n. 9, p. 820-832, 31 ago. 2023. DOI: 10.1056/NEJMoa2215643. Disponível em: https://www.nejm.org/doi/10.1056/NEJMoa2215643. Acesso em: 7 set. 2025.
WILLIAMS, Thomas N.; THEIN, Swee Lay. Sickle Cell Anemia and Its Phenotypes. Annu. Rev. Genom. Hum. Genet.; v. 19, p. 113-147, 2018. Disponível em: https://doi.org/10.1146%2Fannurev-genom-083117-021320. Acesso em: 06 jun. 2023.
ZAGO, M. A. et al. Hematologia: fundamentos e prática. 1. ed. rev. e atualizada. São Paulo: Atheneu, 2004. Acesso em: 01 out. de 2022
1Graduanda em Biomedicina no Centro Universitário Mauricio de Nassau: 40086965@sempreunesc.com.br
2Graduando em Biomedicina no Centro Universitário Maurício de Nassau:
40087528@sempreunesc.com.br
3Mestre em Ciências Ambientais; Graduada em Biomedicina; Docente do Curso de Biomedicina do Centro Universitário Maurício de Nassau de Cacoal E-mail: 400100820@prof.sempreunifacimed.com.br