AUTISM SPECTRUM DISORDER IN A PATIENT WITH NEUROFIBROMATOSIS TYPE 1: CASE REPORT
REGISTRO DOI: 10.69849/revistaft/ra10202510071307
Vanessa Sores de Araújo1; Amanda Castro Nagato2; Ana Laura de Moura Silveira3; Bruna Santana Regattieri De Biase4; Hadassa Motta de Paula Mariano5; Izadora Sant’ana Barrozo de Siqueira6; Jacqueline Moraes Gomes7; Micaele Cristina Rodrigues8; Nathalia Cristine Alves do Nascimento9; Marc Alexandre Duarte Gigonzac10
Resumo
O Transtorno do Espectro Autista (TEA) é um transtorno do neurodesenvolvimento caracterizado por deficiências na comunicação social, interesses restritos e comportamentos repetitivos. O TEA frequentemente coexiste com outros problemas de saúde mental e física. Nos EUA, em 2020, estima-se que 1 em 36 crianças foram identificadas com autismo, e no Brasil, isso equivaleria a cerca de 4.87 milhões de pessoas com o transtorno. O TEA possui uma base genética complexa, dividindo-se em TEA sindrômico (causado por mutações em um único gene) e TEA não sindrômico (associado a vários genes afetados). Estudos com gêmeos mostram um forte componente genético ligado ao autismo, mas também há influências ambientais. A Neurofibromatose Tipo 1 (NF1) é uma condição genética que aumenta o risco de autismo e outros problemas neurológicos. Ela é causada por mutações no gene NF1 e pode se manifestar devido a neomutações em até 50% dos pacientes, não exigindo histórico familiar. Pessoas com NF1 têm uma maior probabilidade de apresentar traços autistas, além de riscos de desenvolver tumores e outros sintomas físicos, como manchas de café com leite na pele. O diagnóstico precoce é fundamental para ambas as condições, permitindo a implementação de planos de cuidados adequados para melhorar a qualidade de vida dos pacientes. Este estudo relata o caso de um paciente com NF1 e autismo na infância, buscando correlações com a literatura sobre diagnóstico, genética e manifestações clínicas.
Palavras-chave: Transtorno do Espectro Autista; Neurofibromatose 1.
1 INTRODUÇÃO
O Transtorno do Espectro Autista (TEA) é um transtorno do neurodesenvolvimento caracterizado por deficiências na comunicação social, interesses restritos e comportamentos repetitivos, segundo o Manual Diagnóstico e Estatístico de Transtornos Mentais (DSM-5), de 2013. Esses sintomas estão presentes desde o início da infância e resultam em desafios sociais generalizados, além de comprometimento na qualidade de vida do paciente. O autismo é frequentemente associado a distúrbios médicos, como Transtorno de Déficit de Atenção e Hiperatividade (TDAH), ansiedade, problemas alimentares, epilepsia, alterações no humor e no sono 1, 2, 3.
De acordo com o relatório mais recente dos Centers for Disease Control and Prevention (CDC), dos EUA, estimou-se que, em 2020, 1 em 36 crianças foi identificada com TEA. Apesar de muitos países não disporem desses dados tão bem documentados — incluindo o Brasil —, ao deslocar a prevalência de autismo encontrada nos EUA (equivalente a 2,3% da população) para a população brasileira, no mesmo período, resultaria em aproximadamente 4.870.380 pessoas com o transtorno no país 4, 5.
Do ponto de vista genético, o Transtorno do Espectro Autista é uma condição que pode ser classicamente categorizada em dois tipos: TEA sindrômico e não sindrômico. O TEA sindrômico é causado por mutações em um determinado gene (modelo monogênico) e se manifesta no contexto de síndromes neurológicas específicas. O TEA não sindrômico está associado a diversos genes afetados (modelo poligênico) 3.
A hereditariedade do TEA não sindrômico foi constatada com base em uma grande diferença nas taxas de concordância, ou estimativas de herdabilidade, entre gêmeos monozigóticos e dizigóticos: gêmeos monozigóticos têm taxas de concordância mais altas para o autismo (variando de 70% a 90%) do que gêmeos dizigóticos (variando de 0% a 30%). Nessa arquitetura genética heterogênea, as formas sindrômicas de autismo, causadas por mutações altamente penetrantes em um único gene, representam apenas uma minoria dos casos de TEA. Os autismos sindrômicos se manifestam no contexto de síndromes neurológicas, como a Síndrome do X Frágil, a Neurofibromatose Tipo 1, a Síndrome de Angelman e o Complexo de Esclerose Tuberosa 3, 10.
A Neurofibromatose Tipo 1 (NF1), ou doença de von Recklinghausen, é uma condição genética autossômica dominante causada por variantes patogênicas no gene NF1 (17q11.2). Possui incidência estimada entre 1:2.500 e 1:3.000 indivíduos, independentemente de sexo ou etnia. Em até 50% dos pacientes, não há história familiar conhecida — ou seja, a doença se manifesta devido a uma neomutação 11, 12.
Um corpo crescente da literatura tem indicado uma carga elevada de traços autistas em pessoas com NF1. Estudos que empregam instrumentos de diagnóstico bem estabelecidos também sugerem que a prevalência de TEA em crianças com NF1 varia entre 11% e 26%, significativamente superior à prevalência de autismo relatada na população em geral 13.
Além de maior predisposição a um quadro autístico, especialmente no domínio das habilidades de comunicação, indivíduos com Neurofibromatose Tipo 1 apresentam risco aumentado de problemas de desenvolvimento neurológico, como déficits cognitivos e de aprendizagem, atrasos motores e sintomatologia de TDAH 11, 14.
Pacientes com essa condição genética também apresentam maior risco de desenvolvimento de tumores do sistema nervoso central ou periférico, pois o gene NF1, que atua na supressão tumoral, encontra-se alterado na Neurofibromatose Tipo 1. Anomalias de pigmentação da pele, como manchas café com leite, sardas nas dobras cutâneas, neurofibromas dérmicos, nódulos de Lisch, anormalidades esqueléticas, neurofibromas plexiformes e gliomas da via óptica são frequentes em pessoas com NF1 12, 15, 16.
Como uma condição monogênica com alta penetrância no Transtorno do Espectro Autista, a Neurofibromatose Tipo 1 apresenta um modelo genético valioso para o avanço da compreensão dos mecanismos neurobiológicos do autismo 13, 17.
O diagnóstico clínico da Neurofibromatose Tipo 1 em uma criança geralmente é inicialmente suspeitado com base em manchas café com leite e pode ser confirmado por meio de teste genético, o qual também é importante para auxiliar no aconselhamento genético e no planejamento familiar 14.
O diagnóstico precoce é fundamental tanto para a Neurofibromatose Tipo 1 quanto para o Transtorno do Espectro Autista, pois permite o estabelecimento de planos de vigilância para questões médicas, de desenvolvimento e comportamentais, garantindo melhor qualidade de vida ao paciente 14.
Nesse sentido, o objetivo deste estudo foi realizar um relato de caso clínico de um paciente diagnosticado com NF1 e autismo na infância, correlacionando-o com a literatura a respeito do diagnóstico, da genética e das manifestações clínicas.
2 RELATO DO CASO
O caso apresentado neste estudo foi avaliado pelo Comitê de Ética em Pesquisa (CEP) do Centro Estadual de Reabilitação e Readaptação Dr. Henrique Santillo (CRER), sob parecer de aprovação nº 2.313.270.
O paciente BLGM, sexo masculino, 5 anos e 1 mês, pardo, natural e procedente de Goiânia (GO), compareceu à primeira consulta em outubro de 2022, acompanhado da mãe, de 35 anos, para aconselhamento genético no CRER, onde funciona o Laboratório de Citogenética Humana e Genética Molecular (Lagene), vinculado à Secretaria Estadual da Saúde de Goiás. Na data da avaliação, o paciente tinha 4 anos e 3 meses e havia sido encaminhado por uma neurologista devido à suspeita de Neurofibromatose Tipo 1 e diagnóstico clínico de Transtorno do Espectro Autista (TEA).
A criança é o primeiro filho de casal não consanguíneo. O bisavô, o avô e o pai paternos possuem diagnóstico de neurofibromatose tipo 1. Além disso, o avô paterno apresenta surdez e epilepsia. Segundo a mãe do paciente, não há histórico familiar de malformações, deficiência intelectual, natimortos ou neomortos.
Ao nascimento, DGM observou a presença de manchas “café com leite” no recém-nascido, “semelhantes às manchas do pai”. Com o crescimento, essas manchas aumentaram em número e tamanho.
O paciente apresentou atraso no desenvolvimento neuropsicomotor: pronunciou as primeiras palavras aos 2 anos e formou as primeiras frases aos 3 anos. Aos 4 anos, sua fala ainda era deficitária, com frases simples, ecolalia e trocas fonêmicas. Sentou-se sem apoio aos 12 meses, andou com apoio aos 13 meses e sem apoio aos 1 ano e 4 meses, porém com certo desequilíbrio e marcha equina (na ponta dos pés).
A mãe relatou que o pobre contato visual do filho foi observado ainda no período de lactente. Desde muito cedo, DGM também notou a pouca interação da criança com as pessoas: era indiferente à comunicação, não apontava para objetos e não atendia quando chamada pelo nome. Ademais, apresentava estereotipias motoras frequentes, aversão ao toque, inflexibilidade cognitiva, seletividade alimentar e dificuldade para iniciar o sono.
Durante o desenvolvimento, o paciente apresentou crises semelhantes a engasgos, iniciadas aos 3 meses de idade, com cianose e perda de consciência. Iniciou acompanhamento neurológico sob suspeita de epilepsia. Com o passar dos meses, os episódios evoluíram para versão lenta da cabeça à direita e hipertonia dos quatro membros, seguidas de clonias predominantes no hemicorpo esquerdo, ocorrendo uma vez ao dia e repetindo-se cerca de três vezes por semana.
Tomografias computadorizadas e ressonância magnética de crânio apresentaram resultados normais. Um eletroencefalograma, entretanto, indicou possível atividade epileptogênica, posteriormente confirmada. Aos 4 anos e 3 meses, mantendo acompanhamento neuropediátrico, o paciente fazia uso de valproato de sódio e risperidona para controle das crises epilépticas.
A mãe relatou que a criança tenta se comunicar por gestos, usa a mão das pessoas para apontar ou conduzi-las até o que deseja, alinha brinquedos ou foca em partes específicas deles (especialmente as rodas), apresenta dificuldade em brincadeiras lúdicas/imaginativas, fixa a atenção em carrinhos e ventiladores, não imita ações de outras pessoas, demonstra resistência a mudanças na rotina, sensibilidade a determinadas texturas e sons altos, além de estereotipias motoras como flapping (bater ou balançar as mãos), bater as mãos nas orelhas, girar, abrir e fechar portas, lamber e cheirar pessoas e alimentos. Também apresenta seletividade alimentar (aceitando apenas alimentos pastosos) e intolerância a glúten e leite.
DGM relatou ainda que o filho é muito agitado, corre constantemente e apresenta baixo limiar à frustração, com acessos de raiva frequentes. Compreende ordens simples, mas tem dificuldade em obedecê-las. Apresenta distúrbio do sono, necessitando de melatonina, prescrita pelo neuropediatra. A mãe negou episódios de auto ou heteroagressividade.
Ao exame físico, observou-se fácies atípica, tórax simétrico e sem deformidades, mãos e pés sem alterações. Constatou-se discreta hipertricose e manchas “café com leite” na pele: uma infraumbilical, medindo 1,5 × 1,5 cm, e outra no abdome posterior direito, medindo 2,0 × 0,5 cm.
Não foram observados neurofibromas nem freckling axilar, e a quantidade de manchas “café com leite” não permitia o fechamento do diagnóstico clínico de neurofibromatose tipo 1. Todavia, o teste genético indicou uma variante provavelmente patogênica, em heterozigose, no gene NF1 (Tabela 1).
A criança deu continuidade ao acompanhamento com o neuropediatra, fonoaudióloga, terapeuta ocupacional, nutricionista, psicóloga, musicoterapeuta e arteterapeuta, visando a prevenção de complicações em seu quadro de saúde e a melhora da sua qualidade de vida.
Tabela 01 – Resultado do teste genético para neurofibromatose.
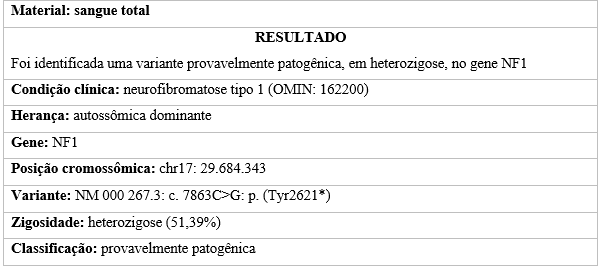
Fonte: elaborada pelos autores.
3 RESULTADOS E DISCUSSÕES
Tendo como base o princípio de que o Transtorno do Espectro Autista (TEA) pode ser categorizado nos tipos sindrômico e não sindrômico, pessoas diagnosticadas com TEA não sindrômico atendem aos critérios do DSM-5, sem outras manifestações somáticas ou neurológicas significativas. Essa forma corresponde à maioria dos indivíduos dentro do espectro. Em contraste, o TEA sindrômico é diagnosticado em indivíduos que atendem aos critérios do DSM-5, mas também apresentam sintomas somáticos ou fenótipos neurológicos adicionais. Esses sintomas podem incluir características dismórficas faciais ou físicas, além de anomalias orgânicas congênitas mais complexas 3, 18.
O diagnóstico clínico do TEA baseia-se em déficits na comunicação social e na presença de comportamentos restritos e repetitivos. Prejuízos na comunicação verbal e não verbal são comuns e impactam o desenvolvimento e a manutenção dos relacionamentos. A intensidade de interesses idiossincráticos ou altamente focados, bem como respostas incomuns a estímulos sensoriais, costuma ser notada desde a primeira infância 1, 19.
A Neurofibromatose Tipo 1 (NF1) é uma doença genética que aumenta o risco de autismo, uma vez que a prevalência de TEA em pessoas com NF1 é mais alta do que na população geral. Isso ocorre porque mutações altamente penetrantes no gene NF1, além de causarem as manifestações típicas da doença, podem desencadear um quadro de autismo no indivíduo acometido 3, 13.
A NF1 apresenta grande variabilidade de expressão clínica, podendo manifestar-se de formas distintas mesmo entre membros de uma mesma família. Entre suas manifestações estão manchas “café com leite”, sardas nas dobras cutâneas, neurofibromas dérmicos, nódulos de Lisch, anormalidades esqueléticas, neurofibromas plexiformes e gliomas da via óptica 16, 20.
Para o diagnóstico clínico de NF1, são necessários pelo menos dois dos sete critérios estabelecidos pela Conferência de Desenvolvimento de Consenso dos Institutos Nacionais de Saúde (NIH), em 1987. Esses critérios incluem: seis ou mais manchas “café com leite”, dois ou mais neurofibromas cutâneos de qualquer tipo ou um neurofibroma plexiforme, sardas axilares ou inguinais, glioma do nervo óptico, dois ou mais nódulos de Lisch, lesões ósseas distintas e pelo menos um familiar de primeiro grau afetado pela doença 12.
Recentemente, o teste genético molecular passou a ser considerado no diagnóstico da NF1. Com uma taxa de sensibilidade de cerca de 95%, é altamente confiável, embora um resultado negativo não exclua completamente a condição 14, 21.
Algumas características da NF1 podem estar presentes desde o nascimento; contudo, a maioria das manifestações surge com o passar do tempo. Assim, quando uma criança preenche os critérios diagnósticos clínicos, o teste genético geralmente não é necessário. Por outro lado, quando há apenas características isoladas, como manchas “café com leite”, o exame genético torna-se fundamental para confirmar a suspeita antes do aparecimento de outras manifestações, como neurofibromas ou gliomas 14.
Nesse contexto, o paciente relatado neste estudo foi encaminhado para avaliação genética por apresentar critérios clínicos sugestivos de NF1, porém insuficientes para confirmação diagnóstica. Apesar de o bisavô paterno, o avô paterno e o pai terem o diagnóstico confirmado, a criança apresentava apenas duas manchas “café com leite”, sem outras alterações observadas. Assim, o teste genético foi essencial para elucidar a condição sindrômica da família.
Embora o fenótipo físico da NF1 possa envolver diversas anormalidades em órgãos e sistemas, os principais desafios relatados por pais e crianças com a doença em ambientes clínicos são de ordem cognitiva, social e comportamental 17.
Estima-se que até 25% das crianças com NF1 atendam aos critérios para TEA, até 45% apresentem sintomas autísticos mais amplos e até 50% recebam diagnóstico de TDAH. BLGM enquadra-se nos 25% de indivíduos com NF1 e autismo. Seus interesses atípicos, como rodas, carrinhos e ventiladores, além da dificuldade em sair da rotina, estereotipias motoras e pouco contato visual, são exemplos característicos do TEA 17.
O comprometimento de linguagem apresentado por BLGM — atraso e dificuldade na fala, ecolalia e trocas fonêmicas — não é específico nem universal ao TEA, embora muitos indivíduos no espectro apresentem atrasos e/ou déficits nessa área 22, 23.
Outra questão relevante neste caso é o diagnóstico de epilepsia. O risco de epilepsia ao longo da vida é aumentado em indivíduos com NF1, variando entre 4% e 14%. Embora lesões focais relacionadas à NF1, como tumores cerebrais, malformações vasculares e corticais, possam explicar cerca de metade dos casos de convulsões, as causas dos casos não estruturais ainda estão sendo investigadas 24. Contudo, entre as pessoas que não apresentam anormalidades estruturais cerebrais, foi observada história familiar de epilepsia em um estudo, sugerindo uma herdabilidade mais complexa do traço epiléptico. Considerando que as tomografias e ressonâncias magnéticas de BLGM não evidenciaram alterações, e que o avô paterno apresenta epilepsia, presume-se que o caso tenha origem genética 24.
Não há cura farmacológica para o TEA nem para a NF1; entretanto, o diagnóstico precoce de ambas as condições traz benefícios significativos aos pacientes. O tratamento medicamentoso é voltado para condições comórbidas, como TDAH, alterações de humor e distúrbios do sono 25.
Uma vez estabelecido o diagnóstico de TEA, os déficits devem ser tratados por meio de abordagem multidisciplinar. Os tratamentos incluem terapias ocupacionais, comportamentais, de fala e lúdicas. As crianças podem receber apoio escolar, incluindo programas de educação especial. Após o diagnóstico de NF1, avaliações oftalmológicas, ortopédicas e neurológicas mais frequentes são recomendadas para permitir intervenções oportunas, especialmente considerando os aspectos tumorais da NF1 25.
4 CONCLUSÃO
Existe uma relação genética entre o Transtorno do Espectro Autista (TEA) e a Neurofibromatose Tipo 1 (NF1), duas condições médicas distintas, mas que podem coexistir em alguns casos. O TEA é um transtorno de neurodesenvolvimento que afeta a comunicação social e o comportamento, enquanto a NF1 é uma doença genética descrita por uma série de manifestações, incluindo manchas café com leite, neurofibromas, déficits cognitivos e de aprendizagem, atrasos motores.
Indivíduos com Neurofibromatose Tipo 1 têm um risco aumentado de desenvolver autismo, o que é corroborado por estudos que identificam uma alta prevalência de traços autistas em pessoas com NF1.
O diagnóstico precoce é de suma importância tanto para o TEA quanto para a NF1, pois isso permite o início de intervenções terapêuticas e de suporte adequadas desde cedo, melhorando a qualidade de vida dos pacientes.
Nesse sentido, o caso clínico apresentado neste estudo ilustra uma criança acometida por ambas as condições, evidenciando suas características clínicas e como uso do teste genético contribuiu para confirmar o diagnóstico de Neurofibromatose Tipo 1.
Em suma, há a necessidade de uma abordagem de equipe multidisciplinar no acompanhamento de crianças com TEA e NF1, incluindo terapias ocupacionais, comportamentais, de fala e lúdicas, bem como avaliações médicas regulares para monitorar complicações associadas à NF1, como tumores. Essa abordagem holística visa melhorar a qualidade de vida e o bem-estar desses pacientes, acompanhando a complexidade das condições e suas interações.
REFERÊNCIAS
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-V). Arlington, VA: American Psychiatric Association, 2013
2. Haebich KM, Pride NA, Walsh KS, Chisholm A, Rouel M, Maier A, et al. Understanding autism spectrum disorder and social functioning in children with neurofibromatosis type 1: protocol for a cross-sectional multimodal study. BMJ Open [Internet]. 2019 Sep 26;9(9):e030601. Available from: https://bmjopen.bmj.com/lookup/doi/10.1136/bmjopen-2019-030601
3. Longo F, Klann E. Reciprocal control of translation and transcription in autism spectrum disorder. EMBO Rep [Internet]. 2021 Jun 4;22(6). Available from: https://www.embopress.org/doi/10.15252/embr.202052110
4. Data & Statistics on Autism Spectrum Disorder [Internet]. Centers for Disease Control and Prevention. 2023 [cited 2023 Sep 30]. Available from: https://www.cdc.gov/ncbddd/autism/data.html
5. IBGE divulga estimativa da população dos municípios para 2020 [Internet]. Agência IBGE Notícias. 2020 [cited 2023 Sep 29]. Available from: https://agenciadenoticias.ibge.gov.br/agencia-sala-de-imprensa/2013-agencia-de-noticias/releases/28668-ibge-divulga-estimativa-da-populacao-dos-municipios-para-2020
6. Genovese A, Butler MG. Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD). Int J Mol Sci [Internet]. 2020 Jul 2;21(13):4726. Available from: https://www.mdpi.com/1422-0067/21/13/4726
7. Alibutud R, Hansali S, Cao X, Zhou A, Mahaganapathy V, Azaro M, et al. Structural Variations Contribute to the Genetic Etiology of Autism Spectrum Disorder and Language Impairments. Int J Mol Sci [Internet]. 2023 Aug 26;24(17):13248. Available from: https://www.mdpi.com/1422-0067/24/17/13248
8. Thapar A, Rutter M. Genetic Advances in Autism. J Autism Dev Disord [Internet]. 2021 Dec 17;51(12):4321–32. Available from: https://link.springer.com/10.1007/s10803-020-04685-z
9. Santos CA dos, Soares Melo HC. A A GENÉTICA ASSOCIADA AOS TRANSTORNOS DO ESPECTRO AUTISTA. Conex Ciência [Internet]. 2018 Oct 10;13(3):68–78. Available from: https://10.253.0.3:49163/index.php/conexaociencia/article/view/756
10. Lubbers K, Stijl EM, Dierckx B, Hagenaar DA, ten Hoopen LW, Legerstee JS, et al. Autism Symptoms in Children and Young Adults With Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex, and Neurofibromatosis Type 1: A Cross-Syndrome Comparison. Front Psychiatry [Internet]. 2022 May 16;13. Available from: https://www.frontiersin.org/articles/10.3389/fpsyt.2022.852208/full
11. Morotti H, Mastel S, Keller K, Barnard RA, Hall T, O’Roak BJ, et al. Autism and attention‐deficit/hyperactivity disorders and symptoms in children with neurofibromatosis type 1. Dev Med Child Neurol [Internet]. 2021 Feb 14;63(2):226–32. Available from: https://onlinelibrary.wiley.com/doi/10.1111/dmcn.14558
12. Mançano AD. Neurofibromatosis type 1. Radiol Bras [Internet]. 2022 Feb;55(1):VII–VIII. Available from: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0100-39842022000100007&tlng=en
13. Chisholm AK, Haebich KM, Pride NA, Walsh KS, Lami F, Ure A, et al. Delineating the autistic phenotype in children with neurofibromatosis type 1. Mol Autism [Internet]. 2022 Dec 4;13(1):3. Available from: https://molecularautism.biomedcentral.com/articles/10.1186/s13229-021-00481-3
14. Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR, et al. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics [Internet]. 2019 May 1;143(5). Available from: https://publications.aap.org/pediatrics/article/143/5/e20190660/37168/Health-Supervision-for-Children-With
15. Tamura R. Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. Int J Mol Sci [Internet]. 2021 May 29;22(11):5850. Available from: https://www.mdpi.com/1422-0067/22/11/5850
16. Kehrer-Sawatzki H, Cooper DN. Challenges in the diagnosis of neurofibromatosis type 1 (NF1) in young children facilitated by means of revised diagnostic criteria including genetic testing for pathogenic NF1 gene variants. Hum Genet [Internet]. 2022 Feb 20;141(2):177–91. Available from: https://link.springer.com/10.1007/s00439-021-02410-z
17. Begum-Ali J, Kolesnik-Taylor A, Quiroz I, Mason L, Garg S, Green J, et al. Early differences in auditory processing relate to Autism Spectrum Disorder traits in infants with Neurofibromatosis Type I. J Neurodev Disord [Internet]. 2021 Dec 28;13(1):22. Available from: https://jneurodevdisorders.biomedcentral.com/articles/10.1186/s11689-021-09364-3
18. Bolbocean C, Andújar FN, McCormack M, Suter B, Holder JL. Health-Related Quality of Life in Pediatric Patients with Syndromic Autism and their Caregivers. J Autism Dev Disord [Internet]. 2022 Mar 3;52(3):1334–45. Available from: https://link.springer.com/10.1007/s10803-021-05030-8
19. Genovese A, Butler MG. The Autism Spectrum: Behavioral, Psychiatric and Genetic Associations. Genes (Basel) [Internet]. 2023 Mar 9;14(3):677. Available from: https://www.mdpi.com/2073-4425/14/3/677
20. Yoshida Y. Neurofibromatosis 1 (von Recklinghausen Disease). Keio J Med [Internet]. 2023;2023-0013-IR. Available from: https://www.jstage.jst.go.jp/article/kjm/advpub/0/advpub_2023-0013-IR/_article
21. Napolitano F, Dell’Aquila M, Terracciano C, Franzese G, Gentile MT, Piluso G, et al. Genotype-Phenotype Correlations in Neurofibromatosis Type 1: Identification of Novel and Recurrent NF1 Gene Variants and Correlations with Neurocognitive Phenotype. Genes (Basel) [Internet]. 2022 Jun 23;13(7):1130. Available from: https://www.mdpi.com/2073-4425/13/7/1130
22. Rosen NE, Lord C, Volkmar FR. The Diagnosis of Autism: From Kanner to DSM-III to DSM-5 and Beyond. J Autism Dev Disord [Internet]. 2021 Dec 24;51(12):4253–70. Available from: https://link.springer.com/10.1007/s10803-021-04904-1
23. Schaeffer J, Abd El-Raziq M, Castroviejo E, Durrleman S, Ferré S, Grama I, et al. Language in autism: domains, profiles and co-occurring conditions. J Neural Transm [Internet]. 2023 Mar 16;130(3):433–57. Available from: https://link.springer.com/10.1007/s00702-023-02592-y
24. Sorrentino U, Bellonzi S, Mozzato C, Brasson V, Toldo I, Parrozzani R, et al. Epilepsy in NF1: Epidemiologic, Genetic, and Clinical Features. A Monocentric Retrospective Study in a Cohort of 784 Patients. Cancers (Basel) [Internet]. 2021 Dec 17;13(24):6336. Available from: https://www.mdpi.com/2072-6694/13/24/6336
25. Al-Dewik NI. Risk factors diagnosis prognosis and treatment of autism. Front Biosci [Internet]. 2020;25(9):4873. Available from: https://imrpress.com/journal/FBL/25/9/10.2741/4873
1 Discente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. E-mail: vanessa_soaresaraujo@hotmail.com
2 Discente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. E-mail: amandacnagato@gmail.com.
3 Discente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. E-mail: anamourassilveira@gmail.com.
4 Discente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. E-mail: brubiasereg@icloud.com.
5 Discente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. E-mail: hadassamdp@gmail.com.
6 Discente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. E-mail: izasiqueira123@gmail.com.
7 Discente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. E-mail: jacqueline.morais67@gmail.com.
8 Discente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. E-mail: micaelecristinarodrigues@gmail.com.
9 Discente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. E-mail: Nathalia_ncan122@hotmail.com.
10 Docente do Curso Superior de Medicina da Pontifícia Universidade Católica de Goiás. Doutor em biotecnologia e biodiversidade pelo programa de Biotecnologia e Biodiversidade da Rede Pró-Centroeste (UnB/UFG). E-mail: m.gigonzac@gmail.com.