REGISTRO DOI: 10.5281/zenodo.7772062
Gilberto Pereira da Cruz
Roberto Macedo Cruz
Orientador: Prof. Me. Arlindo Gonzaga Branco Junior*
Resumo
A ferramenta de edição genética CRISPR possui potencial de tratamento específico para pacientes com fibrose cística, seja essa condição conhecida ou não previamente ao nascimento, devido à característica potencialmente abrangente da técnica de edição genética comparada a outros métodos de edição. Nesse estudo serão compiladas informações sobre os avanços na tecnologia CRISPR e a utilização desta em ensaios clínicos sob a perspectiva dos desafios de implementação da técnica no país, além de comparações desta abordagem de edição genética com estratégias de tratamento estabelecidas no tratamento da fibrose cística.
Palavras-chave: Fibrose cística, CRISPR, edição genética, Terapia genética
Abstract
The CRISPR gene editing tool has specific treatment potential for patients born with cystic fibrosis, whether this condition is known or not prior to delivery, due to its potentially wide range of targets compared to the other types of genetic editing techniques. The formulation of this article was designed to compile information on advances in CRISPR technology and how viable it is today, challenges on the technique and prominent use in laboratories across the country, comparisons of the technique of gene editing and prospects for recovery from fibrosis cystic syndrome associated with other possible healing methods, whether genetic editing or not.
Keywords: cystic fibrosis, CRISPR , genetic editing , genetic therapy
1. INTRODUÇÃO
Ao estudar a ferramenta CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) mais e mais cientistas estão percebendo o potencial ilimitado que essa possui, e no campo médico não é diferente, várias doenças que normalmente seriam mudanças de vida completas para uma pessoa podem ser curadas com uma praticidade inovadora junto à ferramenta (GULEI, BERINDAN-NEAGOE, 2017), a fibrose cística é uma dessas patologias (SANZ et al., 2017). Ao nascer com fibrose cistica um bebe não consegue formular proteínas CFTR que regulam a passagem iônica nas membranas celulares, e com essa deficiência gera problemas que variam desde um suor iônico devido a inabilidade de retenção do sódio até a completa inabilidade de defecar ou até manter o processo respiratório funcionante (BERGERON,CANTIN, 2019).
A ferramenta CRISPR logicamente não é um processo mágico e simples, mas sim uma ferramenta de diversa complexibilidade que pode causar outros aspectos não previstos ainda em modificações genéticas, desta forma, este estudo visa elevar o conhecimento público e explorar todos os outros processos de cura, genéticas ou não, e compará-los à CRISPR para que o leitor possua um conhecimento abrangente e comparativo sobre todas as técnicas, e para isso será utilizada a patologia especificada anteriormente e comparada os seus métodos de tratamento atuais e planejados à técnica CRISPR (UDDIN, RUDIN,SEN, 2020).
1.1 FIBROSE CÍSTICA
Fibrose cística é uma patologia congênita autossômica recessiva, advém de um defeito no gene CFTR, e esse defeito fica localizado no cromossomo 7, e para entender essa patologia é importante entender como a proteína que advém desse gene funciona normalmente no corpo humano. A proteína CFTR é uma proteína de membrana, dependente de ATP, que funciona como canal iônico para o transporte de íons cloreto,seu funcionamento é relativamente básico no corpo o que significa que muitas áreas fisiológicas são dependentes dessa proteína, logo quando defetiva as complicações patológicas irão afetar diversas áreas no corpo humano (ROMMENS et al.,1989).
A proteína tem duas funções específicas, a primeira e mais simples é a sua capacidade de reabsorver íons cloreto do suor, o que normalmente ajuda o corpo humano a manter suas reservas eletrolíticas quando for necessário, e a segunda função é secretar íons cloreto no epitélio externo de órgãos que são geralmente envoltos por muco,esse íon funciona de uma maneira que ao ser secretado atrai o íon de sódio por atração magnética e ao fazê-lo traz junto H20 que normalmente é soluto iônico (LIOU 2019).
Ao atrair H20 no epitélio visceral, essa proteína serve de uma função essencial ao corpo humano que é manter a mucosa visceral hidratada, o que garante o melhor funcionamento de diversas funções no corpo humano, como manter o peristaltismo no trato gastrointestinal funcionando corretamente, manter a função bronquioeslástica normal e garantir que o funcionamento normal pulmonar é mantido, além de outras funções mais obscuras no vas deferens, no cérvix, no pâncreas, no fígado que serão mais discutidas no aspecto patológico deste artigo (LIOU 2019).
Ao entender a fisiologia da fibrose cística, é simples relacionar com as patologias secundárias que advém do defeito genético na proteína CFTR, todas as áreas que normalmente são dependentes da proteína para o funcionamento padrão são agora deficientes do funcionamento padrão e tem risco de desenvolver alguma patologia secundária (WILSCHANSKI 2008).
A fibrose cística leva ao defeito parcial ou completo da formação proteíca CFTR, e tal defeito leva a insuficiência de Íons cloreto na mucosa visceral,além da alta excreção de íons cloreto no suor. Quando a secção de diagnóstico deste artigo for mencionada é importante entender que um dos métodos diagnósticos é utilizar o nível de cloreto no suor,inclusive um método muito usado no passado para o diagnóstico era o gosto salgado do suor de um bebe, geralmente dito como um sinal de morte devido a falta de possibilidade de tratamento na época (BERGERON, CANTIN, 2019).
Os sinais patológicos da fibrose cística vão ser correlacionados com seu uso fisiológico, enquanto o pulmão normalmente tem um muco líquido e útil para lubrificação com seus constantes movimentos, o muco grosso e desidratado de uma pessoa com fibrose cística leva a uma diminuição da capacidade expansionária do pulmão, levando a hipóxia e taquicardia, além do fato de que o acúmulo de muco é uma predisposição enorme para patologias secundárias que normalmente são secundárias a acumulo de muco pulmonar, como pneumonias recorrentes (BERGERON, CANTIN, 2019).
Um dos sinais patognomônicos de um paciente adolescente ou mais velho com fibrose cística não diagnosticada são pneumonias recorrentes (geralmente Pseudomonas aeruginosa em adultos, e Staphylococcus aureus caso mais infantil), também achados são bronquites crônicas em pacientes muito jovens e bronquiectasias sem causas notáveis (SILVA et al., 2019).
Ao prejudicar o processo da mucosa gastrointestinal diversos problemas são criados,entre os comuns estão a má absorção de gorduras levando a problemas relacionando a não absorção de vitaminas lipossolúveis (A,B,D, entre outras), uma possível complicação diabética devido ao dano nas células beta pancreáticas levando ao pâncreas não ser capaz de produzir insulina,a incapacidade parcial ou total de defecação devido a perda da peristalse gastrointestinal entre outros problemas que logo serão discutidos (ESTRADA-VERAS, GRONINGER, 2013).
Como a fibrose cística é uma doença autossômica recessiva é requerido que os dois pais sejam carreadores das falhas genéticas, normalmente heterozigotos, e o padrão é que pacientes homens serem inférteis devido a ausência do vas deferens incapacitando a fertilidade. Mulheres em outro aspecto geralmente são sub-férteis devido a hiperdensidade mucosa vaginal, que é similar ao mecanismo anticoncepcional de progesterona consistente. Um em cada 2500 bebês nascidos serão comprometidos por fibrose cística, é a patologia genética mais comum, logo é um dos diagnósticos bem estabelecidos para serem testados ao nascer o bebe, no brasil o teste é conhecido como “teste do pezinho” que entre suas possibilidades tem o tripsinogênio imunorreativo, que se dado positivo deve ser realizado o teste do suor para confirmar o diagnóstico desse paciente (HULL, KASS, 2000).
Mesmo com todo o apoio necessário atualmente para descobertas ainda assim as dificuldades existentes na vida de um paciente acomprometido por fibrose cistica é consideravel, porem um questionario realizado em 1991 demonstra a vontade de pacientes que eram familiarizados com fibrose cistica, caso tivessem filhos com a patologia, de manter a gravidez até a vida sem considerar o aborto na maioria dos casos, por mais que todos os entrevistados concordassem com o aborto eugênico em caso de morte imediata do feto, 40% aprovariam o aborto caso o bebe morresse antes de chegar aos 5 anos, e poucos iam abortar caso a expectativa fosse 60 anos. Logo, parentes de pessoas que são comprometidas por fibrose cística, que vivem a realidade constantemente, tem como opinião que ainda vale a pena viver com essa patologia (WERTZ et al,. 1991).
1.2 FIBROSE CÍSTICA : Tratamentos paliativos disponíveis
Essencialmente é importante entender que na fibrose cística o paciente sempre teve inúmeros problemas que atualmente são tratados paliativamente, ou seja, quando aparecem no paciente são tratados sem realmente curar o paciente da causa origem,porém a causa principal continua ativa (ESTRADA-VERAS, GRONINGER, 2013). Existem fármacos que possuem capacidade parcial de renovar a função da proteína CFTR como o ivacaftor e o lumacaftor porém esses são limitantes devido a serem restritos ao tratamento de tipos específicos de mutações na fibrose cística e possuírem custos elevados, com valores acima de cem mil reais por ano. É essencial compreender que atualmente os tratamentos são em sua maioria paliativos, portanto é indispensável, por parte do médico, a prática do ensino biopsicossocial às famílias de indivíduos com FC (MIDDLETON et al., 2019).
Atualmente os fármacos usados incluem agentes mucolíticos como N-acetilcisteína ou dornase alfa, que servem para retirar parcialmente o muco grosso presente nas paredes viscerais pulmonares e gástricas de pacientes (HENKE, RATJEN, 2007), também são usado fármacos que são broncodilatadores como albuterol para auxiliar na respiração e perfusão desse paciente,soluções hipertônicas para tentar retirar água das células para o lúmen, azitromicina e AINES como ibuprofeno tentando promover o processo anti-inflamatório e enzimas pancreatitis como insulinas são oferecidas para pacientes demonstrando diabetes secundária à fibrose cística (WALLACE, HALL, KUHN, 1993).
Dentre todos esses medicamentos é importante definir como funciona uso por uso, soluções hipertônicas por exemplo são soluções baratas e simples para uso que se provaram eficientes em testes clínicos de longa data, em suma a solução funciona como um processo que força a água a voltar para fora das células onde ela normalmente estaria,já que o cloro excretado pela proteína CFTR teria a levado junto, e a solução com alto nível de sal tem por osmose a simples hidratação dos tecidos mucosos (DONALDSON et al., 2006). Testes provaram que é uma solução barata,segura e simples de ministrar e que deve ser utilizada junto com broncodilatadores em pacientes para tratamento agudo (ELKINS et al., 2006).
Além de tratamentos medicamentosos existe também a possibilidade clássica de fisioterapia para esse paciente, em que a atenção de um profissional serve para ajudar na limpeza e retirada de secreções nas vias aéreas. técnica é responsável por uma boa parte da melhoria atual de vida que os pacientes atualmente tem. Existem várias técnicas que são usadas porém não é estabelecido qual a técnica mais eficiente nesses pacientes, a técnica de oscilação da parede torácica de alta frequência, que é conhecida como “vest” não teve resultados significativos em comparação à técnica de uso padrão para problemas respiratórios que é a técnica de compressão expiratória positiva, porém demonstrou mais uso de antibióticos nesses pacientes (MCILWAINE et al., 2013).
E ao chegar no tópico de antibióticos é necessário comentar sobre as infecções recorrentes que esses pacientes possuem, devido sua dificuldade em retirar completamente o muco extra presente nas vias aéreas, os pacientes com fibrose cística são clássicos pacientes que possuem infecções recorrentes,e ainda por cima são infecções complicadas como pseudomonas aeruginosa, que eventualmente se estabelecem como infecções crônicas nesses pacientes, em até 60% dos adultos. Por mais que muito seja estabelecido no conhecimento, ainda existem alguns buracos que requerem mais estudos para poder erradicar melhor essa sub patologia (LUND-PALAU et al., 2016).
Nos protocolos de tratamento de antibióticos são várias as opções fornecidas para os pacientes de fibrose cística mas também todos vem com um adendo, tal remédio conseguiu erradicar a população de pseudomonas aeruginosa por 1 ano, 2 anos,etc. Ou seja, é comum esses pacientes entenderem que serão infectados novamente e já estão preparados para voltar para o hospital com sua ficha e todos os medicamentos previamente utilizados. Dos medicamentos estudados a informação dos estudos demonstra que não há diferença entre antibióticos inalados,orais ou a combinação dos mesmos, sendo tobramicina testado em solução de inalação,ciprofloxacino testado em oral e colistina inalada mais ciprofloxacino testado em combinação. todos tiveram sucesso em erradicação temporária do antígeno porém nenhum teve em erradicação completa. estes testes não comprovam que o uso de tal medicamentos aumente a vida média desse indivíduo,porém tal afirmação é principalmente por falta de informação disponível (LANGTON, SMYTH, 2014).
1.3 TÉCNICAS MOLECULARES ATUALMENTE EM ESTUDO APLICADOS A FIBROSE CÍSTICA.
O processo de edição genética não é algo novo a ser considerado, desde os primórdios da sociedade a humanidade vem procurando métodos de alterar o seu ambiente de acordo com suas vontades, porém todos esses métodos que atualmente são disponíveis para estudo não são baratos nem facilmente disponíveis para o uso geral de laboratórios, geralmente bloqueando o acesso de diversos pesquisadores que não possuem o apoio financeiro necessário para atuar, métodos como dedos de zinco, TALENS e mega nucleases são usados atualmente em laboratórios porém com um custo de reprodução relevante (URNOV 2018).
Quando comparado com seus predecessores a técnica CRISPR é uma revolução na disponibilidade dos métodos disponíveis para a tentativa em laboratórios de edição genética,
simplesmente a sua disponibilidade e preço é um dos maiores fatores a se considerar quando pensar em evolução, o fato de que qualquer laboratório simples pode também começar a estudar e pesquisar sobre novas técnicas de edições celulares, tanto animal quanto vegetal, é inovador em todas suas formas, e também perigoso quanto a questões éticas (JIANG ,SHEN, 2019).
1.4 TÉCNICA CRISPR
Inicialmente publicada por Jennifer Doudna e Emmanuelle Charpentier em seu artigo “A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity“ A técnica CRISPR vem sido constantemente estudada devido seu potencial enorme em questões de edição genética, e em casos in-vivo humanos um dos processos iniciais lógicos para iniciar seu uso é patologias genéticas que podem ser ligadas a poucas edições genéticas, como a fibrose cística (CHAPENTIER, 2012).
É essencial demonstrar a sua periculosidade e capacidade de destruição,simplesmente enfatizando o quanto que nos como uma sociedade não sabemos até onde vão as possibilidades de edição com CRISPR, tanto que a sua descobridora, Charpentier, demonstrou enormes receios de até revelar a sua existencia devido preceitos eticos de que talvez ditadores ou revolucionarios poderiam usa-la para criar armas de destruição biológica, ou criar o proximo holocausto tentando criar o soldado perfeito para suas guerras (CHAPENTIER, 2012).
O sistema CRISPR que é um acrônimo que, quando traduzido do inglês, significa Repetições Palindrômicas Curtas Agrupadas e Regularmente Inter Espaçadas, consiste em uma parte do DNA bacteriano que normalmente é utilizada para ler e entender certos vírus que a bactéria encontra, para poder armazenar o material genético desse mesmo vírus na bactéria, e depois utilizar esse material como comparação para aniquilar os próximos vírus que essa bactéria encontrar, é composto por repetições de nucleotídeos que se subdivide em dois grupos, um RNA responsável por guiar a proteína ao seu material, e a parte conhecida por Cas-9 (Crispr associated protein 9) que é uma endonuclease que possibilita a abertura e alteração do sítio de DNA específico (REDMAN et al., 2016).
Das possíveis utilidades da técnica CRISPR, as que serão discutidas aqui principalmente pertencem a parte medicinal, especificamente o tratamento da fibrose cística, junto com algumas menções de outras patologias cujo tratamento com CRISPR tem um potencial de servir como base e inspiração para mais estudos, por exemplo o tratamento de distrofia muscular é um dos usos que está sendo amplamente discutido e estudado, e assim como Fibrose cística, distrofia muscular é uma alteração genética com um componente singular, a mutação DMD em uma mutação de sentido trocado (missense mutation), e já teve seu uso testado com sucesso em ratos no laboratório (ZHANG et al., 2017).
(MAULE, AROSIO, CERESETO,2020) enfatiza que :
A edição do genoma CRISPR é uma tecnologia muito promissora para gerar novas estratégias terapêuticas, bem como novas ferramentas experimentais valiosas para testar terapias para uma ampla variedade de mutações causadoras de Fibrose cística
Com todo o processo histórico da edição genética em mente é notável a razão do quão controverso tem sido esse tópico, porém os benefícios são imensuráveis, simplesmente por ter a capacidade de tratamento de doenças que até agora aterrorizam a humanidade, sendo a fibrose cística uma das maiores causas de doenças genéticas autossômicas recessivas em caucasianos, sendo que há 100 anos atrás raramente uma pessoa sobreviveria após os 5 anos,e até 1970 era raro a taxa de sobrevivência até a adolescência (SANDERS, FINK, 2016).
Muito do progresso curativo que tem atualmente sendo desenvolvido para a fibrose cística é devido a fundação de fibrose cística (cystic fibrosis foundation – cff.org) que são um grupo enorme de trabalhadores da área da saúde, muitos deles com parentes ou eles mesmos são comprometidos por fibrose cística, atualmente na busca por uma melhoria, imediata ou duradoura. Essa fundação mundial notou que CRISPR poderia ser a chave de todo esse sofrimento que compromete milhões de pessoas e investiu muitos recursos nesse foco, além de criarem um processo simples e extremamente proficiente de cadastro de todos os pacientes com fibrose cística, para garantir que estudos feitos são facilmente distribuídos e estabelecidos em seus pacientes (SANDERS, FINK, 2016).
1.5 JUSTIFICATIVA
Por ser uma técnica tão recente, a CRISPR está constantemente mudando e é necessário constantes revisões e artigos para poder manter o conhecimento sobre tal técnica atualizado, revisões sistemáticas claras e objetivas, além de rica em detalhes como esse projeto visa ser, servem como um auxílio para os estudantes que necessitam ler uma compilação sobre o tópico.
O potencial médico com essa técnica é complexo e tênue, devido ao histórico de uso dela não ser o mais ético possível, logo é necessário garantir que todos os ensaios clínicos atualmente ocorrendo estejam seguindo protocolos éticos, visando que a segurança no uso humano em edição genética seja a maior prioridade.
2. MATERIAIS E MÉTODOS
“CRISPR é uma técnica eficaz na terapia de fibrose cística?” Com essa questão em mente será realizado o início de uma revisão sistemática. Os dados são secundários, basearam-se no banco de dados do PUBMED e Tripdatabase
Publicações com “CRISPR cystic fibrosis” ou “CRISPR fibrose cística” em títulos, resumos e palavras-chave serão contados entre os anos de 2019 e 2022. Foram considerados os artigos escritos em Português e Inglês. Revisões bibliográficas, editoriais ou comunicações não foram incluídos na Revisão Sistemática.
Por relevância serão considerados os artigos das categorias e nessa ordem: Terapia genética, medicina, genética, biomedicina, enfermagem
Na busca inicial, 139 trabalhos foram encontrados,sendo os 139 artigos publicados em inglês ou português,a partir do refinamento por tempo identificou-se 95 artigos,selecionou-se os artigos na íntegra, aplicou-se o seguinte teste de relevância em que pelo menos uma das afirmativas é verdadeira. O estudo aborda CRISPR como um tratamento para fibrose cística? O estudo aborda modelos possíveis tratamentos de fibrose cística comparando-os com a técnica CRISPR?
Dos 95 artigos aos quais foram selecionados 90 pelo título e resumo. A aplicação dos testes de relevância resultou em 74 artigos que serão novamente analisados pela leitura na íntegra.
2.1 METODOLOGIA
A pesquisa a ser abordada é uma pesquisa por revisão sistemática utilizando estudos clínicos sobre a utilização de CRISPR-cas9 em processos laboratoriais procurando viabilizar o seu uso em casos humanos, sua fonte é de fontes de dados com foco completo na técnica crispr como a genomecrispr.org que são grupos focados em publicar o máximo de informação relevante possível sobre essa técnica.
O estudo terá como foco testes feitos em pacientes com fibrose cística em associação com a ferramenta CRISPR. Todos os dados analisados serão retirados do site: clinicaltrials.gov, tendo como base de pesquisa palavras chaves com enfoque em “CRISPR”, “CFTR” E “CRISPR CFTR”.
3. OBJETIVOS
3.1 OBJETIVO GERAL:
Analisar a atual situação e frequência do uso da tecnologia de CRISPR em relação à outras abordagens moleculares no tratamento de fibrose cística.
3.2 OBJETIVOS ESPECÍFICOS:
- Contextualizar os tratamentos paliativos disponíveis para fibrose cística;
- Relatar as diferentes abordagens de edição genética usadas no tratamento da fibrose cística através de ensaios clínicos;
- Pesquisar os ensaios clínicos para o tratamento de Fibrose cística utilizando CRISPR e outras ferramentas de edição genética;
- Analisar o estado atual dos ensaios clínicos atualmente disponíveis em relação a CRISPR e Fibrose cística individualmente e juntos.
- Contemplar outras patologias que atualmente estão sendo pesquisadas com a estratégia de edição genética por CRISPR
4. RESULTADOS
Pesquisou-se sobre todos os ensaios clínicos disponíveis no clinical trials com a palavra chave “CFTR”, a palavra chave “CRISPR” e as duas palavras juntas “CRISPR CFTR”, na primeira pesquisa, “CFTR”, foram encontrados 149 resultados, tendo 48 estudos observacionais, os quais foram excluídos, sobrando 101 resultados, dos quais estão divididos em diversas categorias,sendo elas genericamente agrupadas nas seguintes divisões: Terapia moduladora de CFTR; Diagnóstico; Pesquisa de exercício físico; Mucoativo; Edição genética; terapia mRNA; Medicamentos comuns; Pancreatite; Plantas medicinais Os estados de conclusão dos estudos de CFTR também foram levados em conta e listados nas seguintes categorias: Retirado; ainda não recrutando; Ativo, sem recrutamento; Cancelado; Completo; Desconhecido; Recrutando; Suspendido
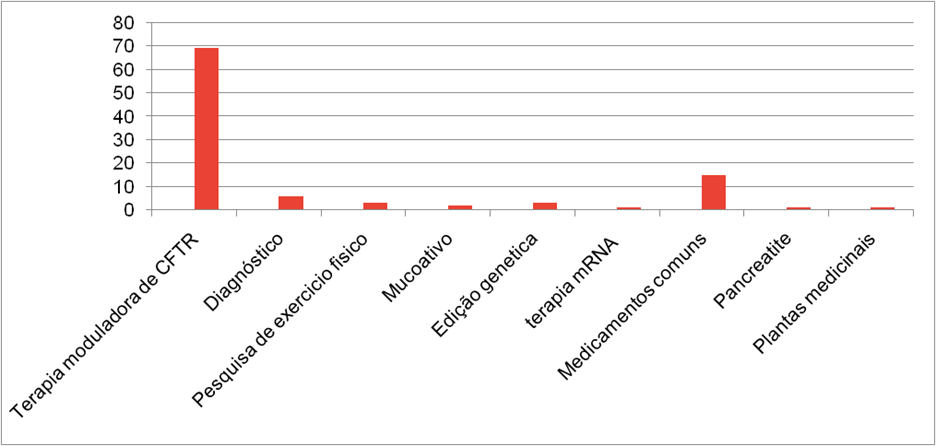
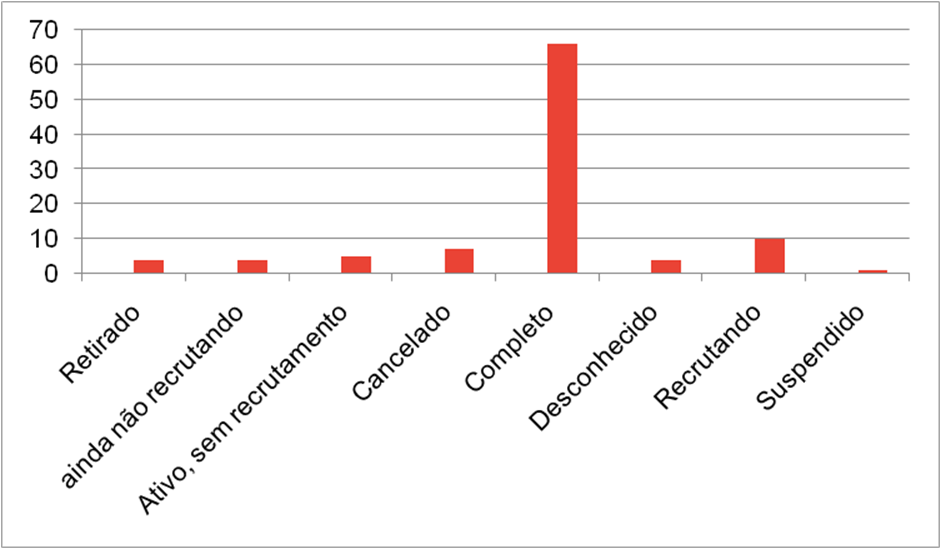
Na Segunda pesquisa “CRISPR” foram encontrados 49 estudos, sendo 7 de origem observacional, logo descartados sobrando 42 resultados, dos quais estão divididos em diversas categorias,sendo elas genericamente agrupadas nas seguintes divisões: Cancer; COVID-19; Distrofia Muscular; Doenças Hematológicas; Infecção Bacteriana; Infecção Viral; Síndrome de Rett; Tumor.
Os estados de conclusão dos estudos de “CRISPR” também foram levados em conta e listados nas seguintes categorias:Ativo, sem recrutamento; Completo; Ainda não recrutando; Recrutando; Cancelado; Desconhecido; Retirado; Recrutamento por convite.
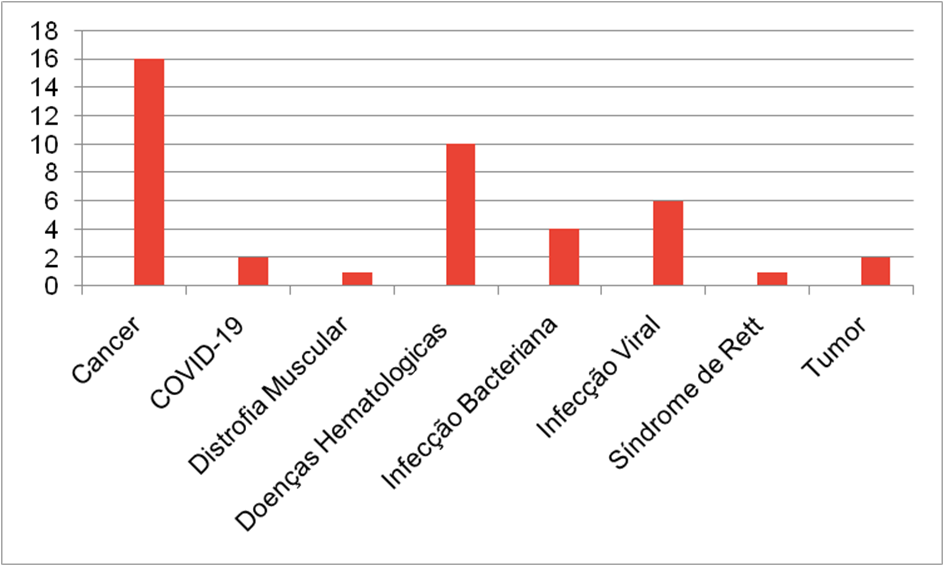
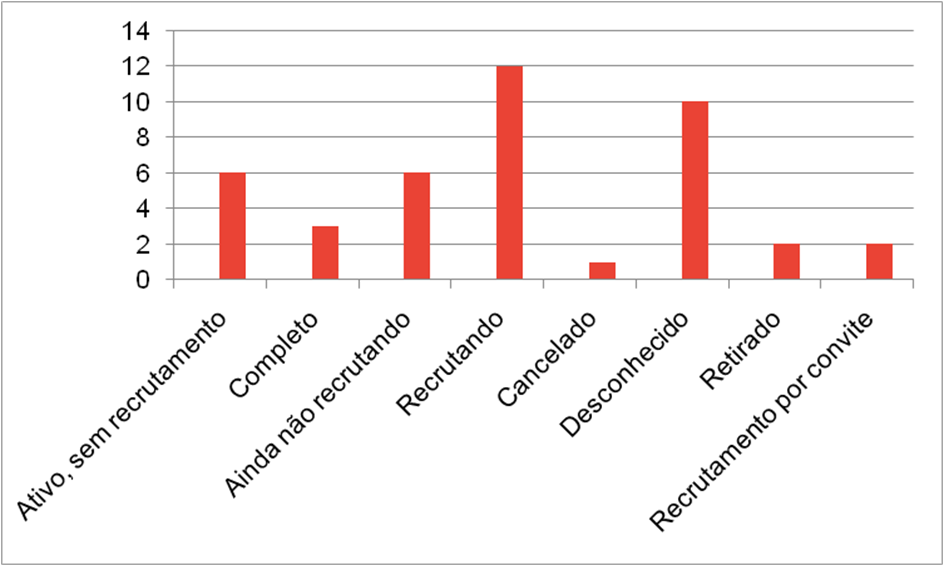
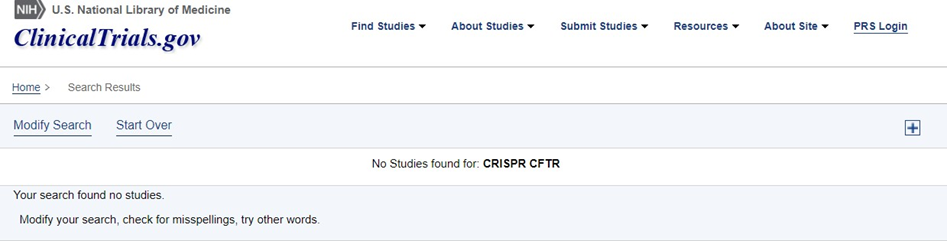
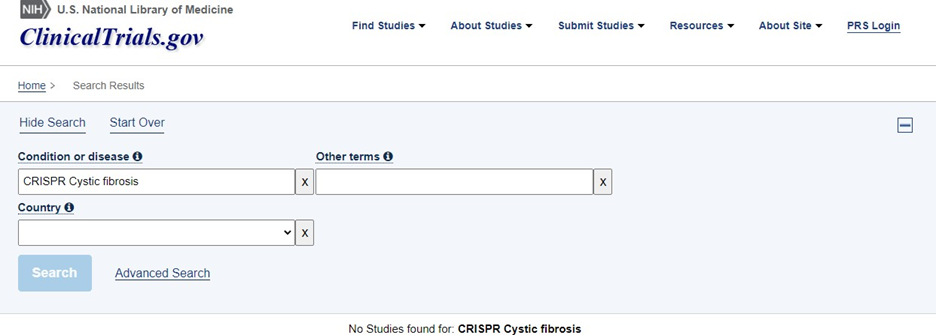
Na terceira pesquisa no clinical trials foi utilizado o termo “CRISPR CFTR” e zero resultados foram encontrados, mesmo com todos os filtros desativados e nenhum critério de seleção ativo, para incrementar e aprofundar foi também adicionado a pesquisa “CRISPR CYSTIC FIBROSIS”, que também resultou em zero estudos encontrados.
6. DISCUSSÃO
A técnica CRISPR atualmente é importante em diversos estudos, como visualizado nos resultados, devido a sua capacidade de aplicação generalizada ser comprovadamente segura. A CRISPR atua de forma universal em todos os indivíduos, sendo uma revolução na edição genética devido à sua precisão, eficiência, versatilidade, acessibilidade e potencial para tratamento de doenças, diferentemente de outras técnicas que precisam ser moduladas de acordo com cada indivíduo. Portanto, enquanto as demais técnicas precisam ser específicas para cada indivíduo, o uso da CRISPR se torna mais interessante devido ao seu amplo uso. (MAULE, ARÓSIO, CERESETO,2020)
Sua capacidade de editar genes com precisão e velocidade sem precedentes abriu novas possibilidades para pesquisa e medicina genética, abrindo novos caminhos para tratamentos e curas de doenças genéticas. Um dos principais pontos positivos na teoria é a especificidade e precisão da CRISPR, porem por mais precisa e teoricamente estável que a técnica seja, estudos não comprovaram essa aplicação na prática. (ZUCCARO, et al., 2020)
Efeitos fora do alvo e mutações indesejadas podem ocorrer durante o processo de edição do gene, levando a potenciais efeitos adversos na saúde do paciente. A segurança e a eficácia do CRISPR para o tratamento da fibrose cística precisam ser cuidadosamente avaliadas em estudos pré-clínicos e clínicos antes que possa ser considerado para uso generalizado, porem os erros encontrados em outos estudos podem ser uma das causas da ausência de ensaios atualmente utilizando a técnica para tratar fibrose cística.
Os focos atuais nos ensaios clínicos foram demonstrados serem questões cancerígenas e doenças hematológicas, como anemia falciforme e beta-talassemias, que são doenças, assim como a fibrose cística, que afetam um gene só no genoma, e que tem potencial de serem curadas com uma simples edição, diminuindo assim o potencial de erro e variação na edição genética.
Os resultados da pesquisa demonstram que os ensaios sobre fibrose cística estão extremamente focados em terapia moduladora da proteína CFTR, utilizando alguma variação do lumacaftor,ivacaftor e tezacaftor, sejam eles juntos ou separados, porém a utilização dessa medicação é algo que vai além da possibilidade da maioria da população, sendo seu custo mensal próximo de 10 salários mínimos, enquanto essa realidade prosseguir o tratamento da fibrose cística continuará sendo algo restrito à poucos, porém provou-se eficaz nos que o tratamento foi testado, logo a indústria farmacêutica e terapêutica aparenta ter pouca motivação para a procura de outras técnicas terapêuticas, sendo a exceção 3 ensaios de edição genéticas que atualmente estão sendo estudados.
Atualmente todos os ensaios de edição genética envolvem a utilização da mesma técnica, a utilização de vectores de adenovírus para procurar realizar a re-funcionalidade da proteína CFTR. Além disso, a fibrose cística é uma doença complexa que envolve vários órgãos e sistemas do corpo. Embora o CRISPR possa potencialmente corrigir a mutação genética subjacente, ele pode não abordar os efeitos secundários da doença, como inflamação, infecção e danos aos órgãos. Portanto, é necessária uma abordagem multifacetada para o tratamento da fibrose cística, que inclua abordar o defeito genético, bem como os efeitos secundários da doença.
7. CONCLUSÃO
Os atuais ensaios clínicos sobre a CRISPR demonstram que existe demanda em sua utilização, todavia, não quando relacionada a fibrose cística, tendo como razão diversas possibilidades, desde razões financeiras motivadas no interesse da indústria farmacêutica pautada no lucro sobre uso vitalicio de medicamentos usados no tratamento da FC, até razões de insegurança em utilizar uma técnica que edita o genoma completo, por se tratar de uma técnica de alto risco devido a efeitos colaterais indesejados.
É necessário tempo para observar se outros tratamentos que estão sendo pesquisados serão eficazes com a técnica CRISPR, e se esse possível sucesso levará a novas pesquisas em áreas atualmente não desejadas, um sucesso por exemplo na erradicação da doença em um paciente com anemia falciforme pode levar a estudos que abram portas para diversas patologias com defeitos similares geneticamente.
Portanto, evidenciou-se que atualmente os ensaios clínicos na fibrose cística tem um viés focado na terapia de modulação da proteína CFTR, porém com uma leve possibilidade da edição genética utilizando adenovírus, no entanto até o presente momento a técnica de CRISPR não tem enfoque algum nos ensaios dessa patologia.
REFERÊNCIAS
GULEI, D., BERINDAN-NEAGOE, I. (2017). CRISPR/Cas9: A Potential Life-Saving Tool. What’s next?. Molecular therapy. Nucleic acids disponível em:<https://doi.org/10.1016/j.omtn.2017.10.013> acesso em 25 de mar de 2022.
BERGERON, C., CANTIN, A. M. (2019). Cystic Fibrosis: Pathophysiology of Lung Disease. Seminars in respiratory and critical care medicine disponível em: <https://doi.org/10.1055/s-0039-1694021> acesso em 25 de mar de 2022.
AYANOĞLU, F. B., ELÇIN, A. E., ELÇIN, Y. M. (2020). Bioethical issues in genome editing by CRISPR-Cas9 technology. Turkish journal of biology disponível em: <https://doi.org/10.3906/biy-1912-52>
UDDIN, F., RUDIN, C. M., SEN, T. (2020). CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Frontiers in oncology disponível em: <https://doi.org/10.3389/fonc.2020.01387> acesso em 25 de mar de 2022
ROMMENS, J. M., IANNUZZI, M. C., KEREM, B., DRUMM, M. L., MELMER, G., DEAN, M., ROZMAHEL, R., COLE, J. L., KENNEDY, D., HIDAKA, N. (1989). Identification of the cystic fibrosis gene: chromosome walking and jumping. Science (New York, N.Y.) disponível em : <https://doi.org/10.1126/science.2772657> acesso em 25 de mar de 2022
SANDERS DB, FINK AK.(2016) Background and Epidemiology. Pediatr Clin North Am. disponível em: <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4967225/> acesso em 03 de jul de 2022.
WAINWRIGHT, C. E., ELBORN, J. S., RAMSEY, B. W., MARIGOWDA, G., HUANG, X., CIPOLLI, M., COLOMBO, C., DAVIES, J. C., DE BOECK, K., FLUME, P. A., KONSTAN, M. W., MCCOLLEY, S. A., MCCOY, K., MCKONE, E. F., MUNCK, A., RATJEN, F., ROWE, S. M., WALTZ, D., BOYLE, M. P., TRAFFIC Study Group, … TRANSPORT Study Group (2015). Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. The New England journal of medicine, disponível em: <https://doi.org/10.1056/NEJMoa1409547> acesso em 24 de mar de 2022.
ROSENSTEIN, B. J., & CUTTING, G. R. (1998). The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. The Journal of pediatrics, disponível em: <https://doi.org/10.1016/s0022-3476(98)70344-0> acesso em 24 de mar de 2022.
DE BOECK, K., & AMARAL, M. D. (2016). Progress in therapies for cystic fibrosis. The Lancet. Respiratory medicine, disponível em: <https://doi.org/10.1016/S2213-2600(16)00023-0> acesso em 24 de mar de 2022.
MAULE G, AROSIO D, CERESETO A. (2020) Gene Therapy for Cystic Fibrosis: Progress and Challenges of Genome Editing. International Journal of Molecular Sciences. disponível em: <https://doi.org/10.3390/ijms21113903> acesso em 24 de mar de 2022.
ATHANAZIO, R. A., SILVA FILHO, L., VERGARA, A. A., RIBEIRO, A. F., RIEDI, C. A., PROCIANOY, E., ADDE, F. V., REIS, F., RIBEIRO, J. D., TORRES, L. A., FUCCIO, M. B., EPIFANIO, M., FIRMIDA, M. C., DAMACENO, N., LUDWIG-NETO, N., MARÓSTICA, P., RACHED, S. Z., MELO, S., Grupo de Trabalho das Diretrizes Brasileiras de Diagnóstico e Tratamento da Fibrose Cística. (2017). Brazilian guidelines for the diagnosis and treatment of cystic fibrosis. Jornal brasileiro de pneumologia : publicacao oficial da Sociedade Brasileira de Pneumologia e Tisilogia, disponível em: <https://doi.org/10.1590/S1806-37562017000000065> acesso em 24 de mar de 2022.
SANZ, D. J., HOLLYWOOD, J. A., SCALLAN, M. F., & HARRISON, P. T. (2017). Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PloS one, disponível em:<https://doi.org/10.1371/journal.pone.0184009> acesso em 24 de mar de 2022.
MIDDLETON, P. G., MALL, M. A., DŘEVÍNEK, P., LANDS, L. C., MCKONE, E. F., POLINENI, D., RAMSEY, B. W., TAYLOR-COUSAR, J. L., TULLIS, E., VERMEULEN, F., MARIGOWDA, G., MCKEE, C. M., MOSKOWITZ, S. M., NAIR, N., SAVAGE, J., SIMARD, C., TIAN, S., WALTZ, D., XUAN, F., ROWE, S. M., … VX17-445-102 Study Group (2019). Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. The New England journal of medicine disponível em: <https://doi.org/10.1056/NEJMoa1908639> acesso em 25 de mar de 2022.
WILSCHANSKI M. (2008). Patterns of gastrointestinal disease associated with mutations of CFTR. Current gastroenterology reports disponível em: <https://doi.org/10.1007/s11894-008-0062-3> acesso em 03 de jul de 2022.
LIOU T. G. (2019). The Clinical Biology of Cystic Fibrosis Transmembrane Regulator Protein: Its Role and Function in Extrapulmonary Disease. Chest, disponível em: <https://doi.org/10.1016/j.chest.2018.10.006> acesso em 25 de mar de 2022.
SILVA FILHO, L. V., FERREIRA, F., REIS, F. J., BRITTO, M. C., LEVY, C. E., CLARK, O., & RIBEIRO, J. D. (2013). Pseudomonas aeruginosa infection in patients with cystic fibrosis: scientific evidence regarding clinical impact, diagnosis, and treatment. Jornal brasileiro de pneumologia : publicacao oficial da Sociedade Brasileira de Pneumologia e Tisilogia disponível em: <https://doi.org/10.1590/S1806-37132013000400015> acesso em 25 de mar de 2022.
KULCZYCKI, L. L., KOSTUCH, M., & BELLANTI, J. A. (2003). A clinical perspective of cystic fibrosis and new genetic findings: relationship of CFTR mutations to genotype-phenotype manifestations. American journal of medical genetics. disponível em: <https://doi.org/10.1002/ajmg.a.10886> acesso em 25 de mar de 2022
ESTRADA-VERAS, J., & GRONINGER, H. (2013). Palliative care for patients with cystic fibrosis. Journal of palliative medicine, disponível em: <https://doi.org/10.1089/jpm.2013.9515> acesso em 25 de mar de 2022
HENKE, M. O., & RATJEN, F. (2007). Mucolytics in cystic fibrosis. Paediatric respiratory reviews,disponível em: <https://doi.org/10.1016/j.prrv.2007.02.009> acesso em 25 de mar de 2022
WALLACE, C. S., HALL, M., KUHN, R. J. (1993). Pharmacologic management of cystic fibrosis. Clinical pharmacy, disponível em: <https://pubmed.ncbi.nlm.nih.gov/8306566/> acesso em 25 de mar de 2022
JINEK, M., CHYLINSKI K, FONFARA I, HAUER M, DOUDNA JA, CHARPENTIER E. A (2012 ) programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. disponível em : <https://pubmed.ncbi.nlm.nih.gov/22745249/> acesso em 03 de jul de 2022.
REDMAN M, KING A, WATSON C, KING D. (2016) What is CRISPR/Cas9? Arch Dis Child Educ Pract Ed. disponível em <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4975809/> acesso em 03 de jul de 2022.
ZHANG, Y., LONG, C., Li, H., MCANALLY, J. R., BASKIN, K. K., SHELTON, J. M., BASSEL-DUBY, R., & OLSON, E. N. (2017). CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Science advances. disponível em : <https://doi.org/10.1126/sciadv.1602814> acesso em 03 de jul de 2022.
URNOV F. D. (2018). Genome Editing B.C. (Before CRISPR): Lasting Lessons from the “Old Testament”. The CRISPR journal disponível em <https://doi.org/10.1089/crispr.2018.29007.fyu> acesso em 03 de jul de 2022.
JIANG S, SHEN QW. (2019) Principles of gene editing techniques and applications in animal husbandry. 3 Biotech disponível em <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6318154/> acesso em 03 de jul de 2022.
MCILWAINE MP, ALARIE N, DAVIDSON GF, LANDS LC, RATJEN F, MILNER R, OWEN B, AGNEW JL. (2013) Long-term multicentre randomised controlled study of high frequency chest wall oscillation versus positive expiratory pressure mask in cystic fibrosis. Thorax. disponível em <https://pubmed.ncbi.nlm.nih.gov/23407019/> acesso em 05 de jul de 2022
ELKINS MR, ROBINSON M, ROSE BR, HARBOUR C, MORIARTY CP, MARKS GB, BELOUSOVA EG, XUAN W, BYE PT. (2006) National Hypertonic Saline in Cystic Fibrosis (NHSCF) Study Group. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. disponível em <https://pubmed.ncbi.nlm.nih.gov/16421364/> acesso em 05 de jul de 2022
DONALDSON SH, BENNETT WD, ZEMAN KL, KNOWLES MR, TARRAN R, BOUCHER RC. (2006) Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. disponível em <https://pubmed.ncbi.nlm.nih.gov/16421365/> acesso em 05 de jul de 2022
LUND-PALAU H, TURNBULL AR, BUSH A, BARDIN E, CAMERON L, SOREN O, WIERRE-GORE N, ALTON EW, BUNDY JG, CONNETT G, FAUST SN, FILLOUX A, FREEMONT P, JONES A, KHOO V, MORALES S, MURPHY R, PABARY R, SIMBO A, SCHELENZ S, TAKATS Z, WEBB J, WILLIAMS HD, DAVIES JC. (2016) Pseudomonas aeruginosa infection in cystic fibrosis: pathophysiological mechanisms and therapeutic approaches. Expert Rev Respir Med. disponível em <https://pubmed.ncbi.nlm.nih.gov/27175979/> acesso em 05 de jul de 2022
Langton Hewer SC, Smyth AR. (2014) Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst Rev. disponível em <https://pubmed.ncbi.nlm.nih.gov/25383937/> acesso em 05 de jul de 2022
Wertz DC, Rosenfield JM, Janes SR, Erbe RW. (1991) Attitudes toward abortion among parents of children with cystic fibrosis. Am J Public Health. disponível em <https://pubmed.ncbi.nlm.nih.gov/1854017/> acesso em 05 de jul de 2022
HULL SC, KASS NE.(2000) Adults with cystic fibrosis and (in)fertility: how has the health care system responded? J Androl. disponível em <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4819317/> acesso em 06 de jul de 2022
Zuccaro, M. V., Xu, J., Mitchell, C., Marin, D., Zimmerman, R., Rana, B., … Egli, D. (2020). Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell. disponível em <https://www.cell.com/cell/fulltext/S0092-8674(20)31389-1#bib37> acesso em 11 de mar de 2023
ORCID: https://orcid.org/0000-0003-4821-8677