REGISTRO DOI:10.5281/zenodo.10647491
1Rafael André Ferreira
2Ludmilla Ferreira de Assis
3Reinaldo Reis Pimentel
4Ana Clara Fernandes de Souza
5Pollyanne Nunes Bonfim Gouvêa
6Fabiane de Carvalho Silva
7Fabio Oliveira de Souza
8Geisiane Barbosa de Bastos
9Murillo Monteiro de Oliveira
10Ricardo Viana Velloso
Resumo: A necessidade urgente do desenvolvimento de novos agentes antimicrobianos para combater a resistência microbiana a antibióticos e antifúngicos é evidenciada pela declaração da Organização Mundial da Saúde (OMS) em 2021, que destaca essa resistência como uma das principais ameaças à saúde global. Projeções indicam que, sem ação adequada, a mortalidade global devido à resistência microbiana atingirá 10 milhões por ano até 2050. A comunidade científica busca estratégias inovadoras, incluindo a síntese de novos compostos com o uso da nanotecnologia, por exemplo, para superar essa crise. O presente trabalho aborda a síntese e atividade biológica de agentes antimicrobianos. A abordagem proposta neste trabalho visa destacar descobertas relevantes relacionadas à síntese de fármacos antimicrobianos, mecanismos de ação, aplicação clínica e avanços científicos. A exploração da evolução da síntese de medicamentos desde as primeiras drogas antimicrobianas até as mais recentes é discutida, enfatizando a importância dos medicamentos mais utilizados na prática clínica. O texto aborda também a fundamentação do uso de antimicrobianos, destacando como alguns microrganismos produzem substâncias antimicrobianas na natureza. É salientado que o uso generalizado e muitas vezes desnecessário de antimicrobianos levou ao surgimento de cepas multirresistentes. Os fundamentos bacteriostáticos e bactericidas dos fármacos são discutidos, enfatizando a escolha entre eles com base no tipo de infecção e no estado imunológico do paciente. Em suma, o presente trabalho destaca a urgência da elaboração de novos agentes antimicrobianos, apresentando estratégias em andamento na comunidade científica e explorando os fundamentos do uso responsável desses medicamentos em meio à crescente resistência microbiana.
Palavras-chave: antimicrobianos, antibacterianos, síntese de medicamentos.
Abstract: The urgent need for new antimicrobial agents to combat microbial resistance to antibiotics and antifungals is underscored by the World Health Organization’s (WHO) declaration in 2021, highlighting resistance as a major global health threat. Projections indicate that, without proper intervention, global mortality due to microbial resistance will reach 10 million per year by 2050. The scientific community is exploring innovative strategies, including the synthesis of new compounds and nanoparticles, to address this crisis. This paper focuses on the synthesis and biological activity of antimicrobial agents. The proposed approach aims to highlight relevant findings related to the synthesis of antimicrobial drugs, mechanisms of action, clinical applications, and scientific advancements. The evolution of drug synthesis from early antimicrobial drugs to the latest ones is discussed, emphasizing the significance of commonly used drugs in clinical practice. The text also addresses the rationale behind antimicrobial use, highlighting how certain microorganisms produce antimicrobial substances in nature. However, widespread and often unnecessary use of antimicrobials has led to the emergence of multi-resistant strains. The bacteriostatic and bactericidal fundamentals of drugs are discussed, emphasizing the choice between them based on the type of infection and the patient’s immune status. In conclusion, this paper underscores the urgency of new antimicrobial agents, presenting ongoing strategies in the scientific community and exploring the fundamentals of responsible antimicrobial use amid growing microbial resistance.
Keywords: antimicrobials, antibacterials, drug synthesis.
1 INTRODUÇÃO
O desenvolvimento de agentes antimicrobianos é urgente e necessário a fim de superar a resistência adquirida dos microrganismos aos antibióticos e antifúngicos existentes. No final de 2021, a Organização Mundial da Saúde (OMS) declarou a resistência microbiana a antibióticos como uma das dez ameaças globais à saúde humana. A Comissão Europeia estima que na União Europeia ocorram 33.000 mortes por ano devido à resistência microbiana a antibióticos, com custos relacionados a cuidados de saúde e perda de produtividade que alcançam 1,5 bilhão de euros (WHO, 2022).
De acordo com previsões, até 2050, se nenhuma ação for tomada, a mortalidade global devido à resistência microbiana a antibióticos atingirá 10 milhões por ano, com custos relacionados de cerca de 1 trilhão de dólares. Esses números claramente destacam a necessidade de novos agentes antimicrobianos eficazes capazes de tratar infecções e superar a resistência de microrganismos (bactérias ou fungos). Ciente disso, a comunidade científica está envolvida no estudo de estratégias para descobrir antimicrobianos eficientes que atuem por meio de mecanismos diferentes daqueles atualmente utilizados no combate a infecções e na superação da resistência microbiana adquirida (WHO, 2023; MARINESCU M., 2021).
Uma das estratégias está focada na síntese de novos compostos, sejam eles de coordenação ou orgânicos, e na preparação de nanopartículas a partir da avaliação de suas propriedades antimicrobianas correspondentes. Exemplos de cada uma dessas abordagens constam no Tópico Especial “Síntese e Atividade Biológica de Agentes Antimicrobianos” na revista Antibiotics da MDPI, que inclui dois artigos de revisão e nove artigos de pesquisa sobre a síntese de compostos orgânicos ou de coordenação e nanopartículas, sejam elas simples ou revestidas (TREIBER L. et al., 2021; PINHEIRO M. et al., 2021).
Nesse sentido se faz necessária a presente abordagem a qual tem por principal objetivo situar os achados relevantes em relação à síntese de fármacos antimicrobianos, os mecanismos de ação e aplicação clínica, e importantes aspectos que ressaltam a relevância dos medicamentos sintéticos no cenário terapêutico, além dos avanços científicos convergentes com as possibilidades de melhorias nas tecnologias de produção de medicamentos antimicrobianos.
Destaque-se, por fim, a exploração da evolução da síntese de medicamentos desde a introdução das primeiras drogas antimicrobianas até as mais atuais. Discutiremos também a importância dos medicamentos antimicrobianos mais utilizados na prática clínica, focando na sua estrutura, função e características mais relevantes.
2 FUNDAMENTAÇÃO DA SÍNTESE DE ANTIMICROBIANOS
Na natureza, alguns microrganismos produzem substâncias que inibem ou matam outros microrganismos que, de outra forma, poderiam competir pelos mesmos recursos. Os seres humanos têm explorado com sucesso essas habilidades, usando organismos para produzir em massa substâncias que podem ser usadas como antimicrobianos (WAINWRIGHT V., 1989).
Desde sua descoberta, os antimicrobianos têm salvado inúmeras vidas e continuam sendo uma ferramenta essencial para o tratamento e controle de doenças infecciosas. No entanto, seu uso generalizado e muitas vezes desnecessário teve um efeito colateral não intencional: o surgimento de cepas microbianas multirresistentes a múltiplos medicamentos.Assim, discutiremos como os antimicrobianos funcionam, por que os microrganismos desenvolvem resistência e o que os profissionais de saúde podem fazer para incentivar o uso responsável dos antimicrobianos (BALTZ M., 2007).
2.1 Fundamentos bacteriostáticos e bactericidas dos fármacos.
Os medicamentos antibacterianos podem ser tanto bacteriostáticos quanto bactericidas em suas interações com as bactérias-alvo. Os medicamentos bacteriostáticos causam uma inibição reversível do crescimento, com o crescimento bacteriano reiniciando após a eliminação do medicamento. Em contraste, os medicamentos bactericidas matam as bactérias-alvo. A decisão de usar um medicamento bacteriostático ou bactericida depende do tipo de infecção e do estado imunológico do paciente. Em um paciente com defesas imunológicas fortes, tanto medicamentos bacteriostáticos quanto bactericidas podem ser eficazes para alcançar a cura clínica. No entanto, quando um paciente está imunocomprometido, um medicamento bactericida é essencial para o tratamento bem-sucedido de infecções. Independentemente do estado imunológico do paciente, infecções que representam ameaça à vida, como a endocardite aguda, por exemplo, exigem o uso de um medicamento bactericida (NELSON M.L., 2010).
2.2 Fundamentos do espectro de atividade dos fármacos antimicrobianos
Sobre o espectro de atividade de um medicamento antibacteriano que se relaciona com a diversidade das bactérias alvo, podemos afirmar que:
Um antimicrobiano de espectro estreito direciona-se apenas a subconjuntos específicos de patógenos bacterianos. Por exemplo, alguns medicamentos de espectro estreito atacam apenas as bactérias gram-positivas, enquanto outros visam apenas as bactérias gram-negativas. Se o patógeno que causa uma infecção foi identificado, é melhor usar um antimicrobiano de espectro estreito para minimizar os danos colaterais à microbiota normal (WAINWRIGHT V., 1989).
Um antimicrobiano de amplo espectro interage com uma grande variedade de patógenos bacterianos, incluindo espécies gram-positivas e gram-negativas, sendo frequentemente usado como terapia empírica para cobrir uma ampla gama de patógenos em potencial enquanto aguarda a identificação laboratorial do patógeno infeccioso. Antimicrobianos de amplo espectro também são usados em infecções polimicrobianas (infecções mistas com várias espécies bacterianas) ou como prevenção profilática de infecções em cirurgias/procedimentos invasivos. Por fim, antimicrobianos de amplo espectro podem ser selecionados para tratar uma infecção quando um medicamento de espectro estreito falha devido ao desenvolvimento de resistência do patógeno-alvo (VERMA S. et al., 2008).
A via de administração, o método utilizado para introduzir um medicamento no corpo, também é uma consideração importante para a terapia medicamentosa. Medicamentos que podem ser administrados por via oral geralmente são preferidos, pois os pacientes podem tomá-los mais convenientemente em casa. No entanto, alguns medicamentos não são facilmente absorvidos do trato gastrointestinal (TGI) para a corrente sanguínea. Esses medicamentos são frequentemente úteis no tratamento de doenças do trato intestinal, como o tratamento de tênias com niclosamida, ou na descontaminação do intestino, como no caso da colistina. Alguns medicamentos que não são facilmente absorvidos, como bacitracina, polimixina e vários antifúngicos, estão disponíveis como preparações tópicas para o tratamento de infecções superficiais na pele (BERDY J., 2005).
Às vezes, os pacientes podem não ser capazes de tomar medicamentos por via oral inicialmente devido à sua condição (por exemplo, vômitos, intubação, intolerância gástrica). Quando isso ocorre ou quando um medicamento escolhido não é absorvido no trato gastrointestinal, a administração do medicamento por via parenteral (injeção intravenosa ou intramuscular) é preferida e normalmente é realizada em ambientes de cuidados de saúde. Para a maioria dos medicamentos, os níveis plasmáticos alcançados pela administração intravenosa são substancialmente mais elevados do que os níveis alcançados pela administração oral ou intramuscular, e isso também pode ser uma consideração importante ao escolher a via de administração para o tratamento de uma infecção (figura 01) (BALTZ M., 2007).
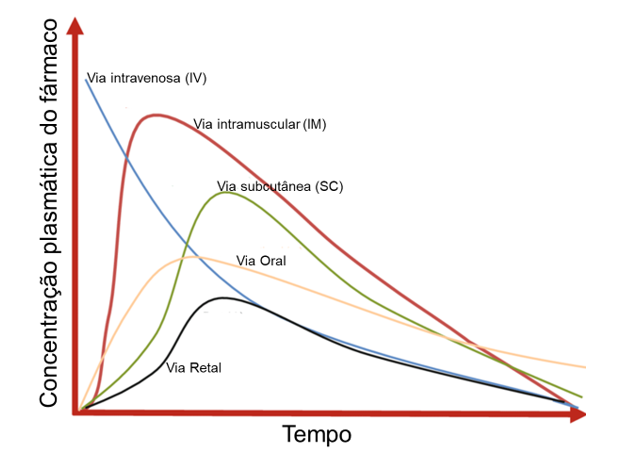
Figura 01: No decorrer do desenvolvimento de novos medicamentos, são conduzidos estudos de biodisponibilidade comparativa. Essa análise comparativa dos resultados permite identificar qual formulação demonstra o perfil mais vantajoso em termos de eficácia e segurança, e aponta características de biodisponibilidade em relação às via de administração. Assim, é possível observar as curvas de concentração plasmática de um determinado fármaco em função do tempo diante de três vias de administração diferentes (oral, intramuscular e intravenosa) o qual é administrado em doses únicas iguais a um mesmo indivíduo sadio em dias diferentes. Adaptado de MARTÍNEZ MF & QUIÑONES LA (2018). Disponível em https://link.springer.com/chapter/10.1007/978-3-319-99593-9_8. Acesso em 02/02/2024).
2.3 Fundamentos das interações medicamentosas de fármacos antimicrobianos
Para o tratamento ideal de algumas infecções, dois medicamentos antibacterianos podem ser administrados juntos para proporcionar uma interação sinérgica que é melhor do que a eficácia de qualquer um dos medicamentos isoladamente. Um exemplo clássico de combinações sinérgicas é o trimetoprim e sulfametoxazol (Bactrim F). Individualmente, esses dois medicamentos fornecem apenas inibição bacteriostática do crescimento bacteriano, mas juntos, os medicamentos são bactericidas (FALAGAS M.E. & KARAGEORGOPOULOS D.E., 2010).
Enquanto as interações medicamentosas sinérgicas beneficiam o paciente, as interações antagonistas produzem efeitos prejudiciais. A antagonismo pode ocorrer entre dois antimicrobianos ou entre antimicrobianos e não-antimicrobianos usados para tratar outras condições. Os efeitos variam dependendo dos medicamentos envolvidos, mas as interações antagonistas podem causar perda da atividade do medicamento, diminuição dos níveis terapêuticos devido ao aumento do metabolismo e eliminação, ou aumento do potencial de toxicidade devido à diminuição do metabolismo e eliminação (DICKINSON B.D. et al., 2001).
Como exemplo, alguns agentes antibacterianos são absorvidos de forma mais eficaz no ambiente ácido do estômago. Se um paciente tomar antiácidos, isso aumentará o pH do estômago e impactará negativamente a absorção desses antimicrobianos, reduzindo sua eficácia no tratamento de uma infecção. Estudos também mostraram uma associação entre o uso de alguns antimicrobianos e a falha de contraceptivos orais (DADGOSTAR P., 2019).
3 METODOLOGIA
O estudo foi elaborado a partir de dados obtidos por meio de revisões bibliográficas. A revisão da literatura foi realizada durante o segundo semestre de 2023 por meio de pesquisa em livros e revistas científicas e em sites de busca como Scielo, PubMed e Google Acadêmico. Ao todo foram selecionados 34 trabalhos para revisão sistemática os quais podem ser consultados no decorrer do texto ou na sessão “referências”. Foram realizadas buscas sobre os principais antimicrobianos com ênfase em seus princípios de ação, características químicas, estruturais e farmacológicas, interações e principais indicações. Foram também destacadas informações como implicações clinicas e efeitos adversos.
4 RESULTADOS E DISCUSSÕES
4.1 Inibidores da biossíntese da parede celular
Várias classes diferentes de agentes antibacterianos bloqueiam etapas na biossíntese de peptidoglicano, tornando as células mais susceptíveis à lise osmótica (Tabela 01). Portanto, os agentes antibacterianos que visam a biossíntese da parede celular são bactericidas em sua ação. Como as células humanas não produzem peptidoglicano, esse modo de ação é um excelente exemplo de toxicidade seletiva (IMPERI F. et al., 2013).
Tabela 01: Exemplo de fármacos antibacterianos, seus mecanismos de ação e principais vias de administração. IM = intramuscular, IV = intravenosa. (Fonte: de autoria própria).
Classes Mecanismo de ação Vias de administração b-lactâmicos Ação bactericida. Atua por inibição da parede celular bacteriana IM, IV, Oral Macrolídeos Agentes bacteriostáticos, inibem a síntese de proteínas, através de ligação às subunidades ribossômicas 50S. Oral, IV Aminoglicosídeos Ação bactericida por causarem alterações em proteínas sintetisadas pela bactéria, ao ligarem-se irreversivelmente aos ribossomas bacterianos. Podem também atuar de forma bacteriostática ao inibirem a síntese de proteínas. IV, IM, Inalatória, Oftálmica e tópica. Quinolonas Agem por inibição do DNA girase, enzima essencial para sobrevivência de uma bactéria, a DNA girase torna a molécula de DNA compacta e biologicamente ativa IV, Oral Sulfonamidas Têm efeito bacteriostático e inibem o metabolismo do ácido fólico, por mecanismo competitivo. Oral Glicopeptideos Inibindo a síntese do peptideoglicano, além de alterar a permeabilidade da membrana citoplasmática e interferir na síntese de RNA citoplasmático IM, IV, Oral Nitroimidazólicos Atua como receptor de elétrons, levando à liberação de compostos tóxicos e radicais livres que atuam no DNA, inativando-o e impedindo a síntese enzimática das bactérias. Oral, IM, IV Lincosamidas Inibem a síntese protéica nos ribossomos, ligando-se a subunidade 50S, sendo bacteriostáticas IV, IM, Polimixinas Interagem com a molécula de polissacarídeo da membrana externa das bactérias gram-negativas, retirando cálcio e magnésio, necessários para a estabilidade da molécula de polissacarídeo. IM ou via intratecal Oxazolidinona Inibição da síntese protéica, mas, em etapa distinta daquela inibida por outros antimicrobiano Oral, IV Tetraciclinas Entram na célula por difusão, em um processo dependente de gasto de energia. Ligam-se, de maneira reversível, à porção 30S do ribossoma, bloqueando a ligação do RNA transportador, impedindo a síntese protéica. Oral, IM, IV
A penicilina, o primeiro antibiótico descoberto, faz parte de várias classes de antibacterianos conhecidos como β-lactâmicos. Esse grupo de compostos inclui as penicilinas, cefalosporinas, monobactâmicos e carbapenêmicos e é caracterizado pela presença de um anel β-lactâmico encontrado na estrutura central da molécula do medicamento (Figura 2). Os antibacterianos β-lactâmicos bloqueiam a ligação cruzada das cadeias peptídicas durante a biossíntese do novo peptidoglicano na parede celular bacteriana. Eles são capazes de bloquear esse processo porque a estrutura β-lactâmica é semelhante à estrutura do componente da subunidade de peptidoglicano reconhecido pela enzima transpeptidase de ligação cruzada, também conhecida como proteína ligante de penicilina (PBP) (LOWY F.D., 2003).
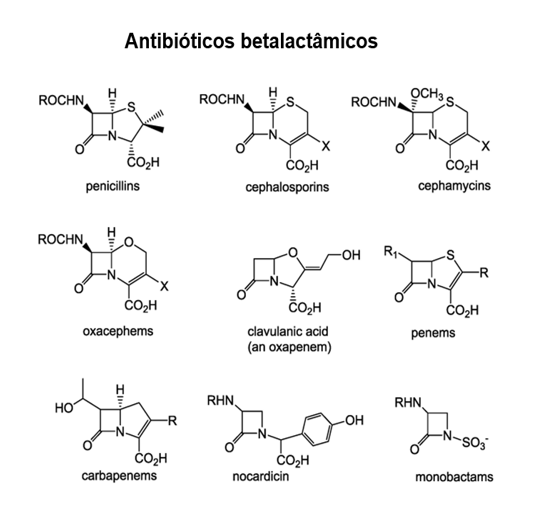
Figura 02: Penicilinas, cefalosporinas, monobactamas e carbapenemas contêm anel β-lactâmico, o local de ataque por enzimas inativadoras de β-lactamase. Embora todas compartilhem o mesmo núcleo, diferentes penicilinas diferem umas das outras na estrutura de seus grupos R. Mudanças químicas nos grupos R proporcionam um aumento no espectro de atividade, estabilidade ácida e resistência à degradação pela β-lactamase (Adaptado de Williams, 1999).
Embora o anel β-lactâmico precise permanecer inalterado para que esses medicamentos mantenham sua atividade antibacteriana, alterações químicas estratégicas nos grupos R permitiram o desenvolvimento de uma ampla variedade de antibióticos β-lactâmicos semissintéticos com maior potência, espectro expandido de atividade e meia-vida mais longa para melhor dosagem, entre outras características (FANORO O. et al., 2021).
A droga vancomicina, um membro de uma classe de compostos chamada glicopeptídeos, foi descoberta na década de 1950 como um antibiótico natural produzido pelo actinomiceto Amycolatopsis orientalis. Semelhante aos β-lactâmicos, a vancomicina inibe a biossíntese da parede celular e é bactericida. No entanto, ao contrário dos β-lactâmicos, a estrutura da vancomicina não é semelhante à das subunidades de peptidoglicano da parede celular e não inativa diretamente as proteínas de ligação à penicilina (GUL S. et al., 2019).
Em vez disso, a vancomicina é uma molécula muito grande e complexa que se liga à extremidade da cadeia peptídica de precursores da parede celular, criando um bloqueio estrutural que impede que as subunidades da parede celular sejam incorporadas na crescente estrutura de N-acetilglucosamina e N-acetilmurâmico (NAM-NAG) do peptidoglicano (transglicosilação) (BERDY J., 2005).
A vancomicina também bloqueia a transpeptidação estruturalmente, sendo bactericida contra patógenos bacterianos gram-positivos (FURUNO J.P. et al., 2014).
4.2 Inibidores da biossíntese de proteínas
4.2.1 Inibidores da síntese de proteínas que se ligam à subunidade 30S
Os aminoglicosídeos são antibióticos antibacterianos grandes e altamente polares que se ligam à subunidade 30S dos ribossomos bacterianos, prejudicando a capacidade de correção do complexo ribossomal. Essa deficiência causa desajustes entre códons e anticódons, resultando na produção de proteínas com aminoácidos incorretos e proteínas encurtadas que se inserem na membrana citoplasmática (LOSEE L. et al., 2015).
Outra classe de compostos antibacterianos que se ligam à subunidade 30S são as tetraciclinas, as quais são bacteriostáticos e inibem a síntese de proteínas bloqueando a associação das moléculas de RNA transportador (tRNAs) com o ribossomo durante a tradução (BALTZ M., 2007).
4.2.2 Inibidores da síntese de proteínas que se ligam à subunidade 50S
Existem várias classes de antibióticos que atuam ligando-se à subunidade 50S dos ribossomos bacterianos. O primeiro macrolídeo foi a eritromicina. Foi isolado em 1952 da Streptomyces erythreus e impede a translocação. Os macrolídeos semissintéticos incluem a azitromicina e a telitromicina. Comparada com a eritromicina, a azitromicina possui um espectro de atividade mais amplo, menos efeitos colaterais e uma meia-vida significativamente mais longa (1,5 horas para eritromicina versus 68 horas para azitromicina), o que permite uma dosagem diária única e um curso de terapia curto de 3 dias (ou seja, a formulação Zpac) para a maioria das infecções (VERMA S., SINGH S.P., 2008).
Os lincosamidas incluem a lincomicina, produzida naturalmente, e a clindamicina, semissintética. Embora estruturalmente distintas dos macrolídeos, as lincosamidas são semelhantes em seu modo de ação aos macrolídeos, ligando-se à subunidade 50S do ribossomo e impedindo a formação de ligações peptídicas. As lincosamidas são especialmente ativas contra infecções estreptocócicas e estafilocócicas (SHATAN A.B. et al.,2021)
As oxazolidinonas, incluindo a linezolida, são uma nova classe de inibidores sintéticos de síntese de proteínas de amplo espectro que se ligam à subunidade 50S dos ribossomos de bactérias gram-positivas e gram-negativas. No entanto, seu mecanismo de ação parece um pouco diferente daquele dos outros inibidores de síntese de proteínas que se ligam à subunidade 50S discutidos anteriormente. Em vez disso, eles parecem interferir na formação do complexo de iniciação (associação da subunidade 50S, subunidade 30S e outros fatores) para a tradução e impedem a translocação da proteína em crescimento do local A do ribossomo para o local P (TORTELLA G. et al., 2021).
4.3 Inibidores da função de membrana
Um pequeno grupo de antibacterianos tem como alvo a membrana bacteriana como seu modo de ação. As polimixinas são antibióticos polipeptídicos naturais que foram descobertos pela primeira vez em 1947 como produtos de Bacillus polymyxa; apenas a polimixina B e a polimixina E (colistina) foram usadas clinicamente. O antibacteriano daptomicina é um lipopeptídeo cíclico produzido pelo Streptomyces roseosporus que parece funcionar de maneira semelhante às polimixinas, inserindo-se na membrana celular bacteriana e a desestabilizando (TREIBER L. et al., 2021)
No entanto, ao contrário da polimixina B e da colistina, que têm como alvo apenas bactérias gram-negativas, a daptomicina tem como alvo especificamente as bactérias gram-positivas. Ela é tipicamente administrada por via intravenosa e parece ser bem tolerada, mostrando toxicidade reversível nos músculos esqueléticos (VAZ, L.E., et al., 2015).
4.4 Inibidores da síntese de ácidos nucleicos
Alguns medicamentos antibacterianos funcionam inibindo a síntese de ácidos nucleicos. Por exemplo, o metronidazol é um membro semissintético da família dos nitroimidazóis e também é um antiprotozoário. Ele interfere na replicação do DNA nas células-alvo. O medicamento rifampicina é um membro semissintético da família dos rifamicinas e funciona bloqueando a atividade da RNA polimerase nas bactérias. Um membro da família das quinolonas, um grupo de antimicrobianos sintéticos, é o ácido nalidíxico (THE WHITE HOUSE, 2015).
Ele foi descoberto em 1962 como subproduto durante a síntese da cloroquina, um medicamento antimalárico. O ácido nalidíxico inibe seletivamente a atividade da DNA girase bacteriana, bloqueando a replicação do DNA. Modificações químicas na estrutura original das quinolonas resultaram na produção de fluoroquinolonas, como ciprofloxacina e levofloxacina, que também inibem a atividade da DNA girase. Ciprofloxacina e levofloxacina são eficazes contra um amplo espectro de bactérias gram-positivas ou gram-negativas e estão entre os antibióticos mais prescritos para tratar uma ampla gama de infecções, incluindo infecções do trato urinário, infecções respiratórias, infecções abdominais e infecções de pele (MARINESCU M., 2021).
4.5 Inibidores de vias Metabólicas
Alguns medicamentos sintéticos controlam infecções bacterianas atuando como antimetabólitos, inibidores competitivos de enzimas metabólicas bacterianas. As sulfonamidas (sulfas) são os agentes antibacterianos sintéticos mais antigos e são análogos estruturais do ácido para-aminobenzóico (PABA), um intermediário inicial na síntese do ácido fólico. Trimetoprima é um composto antimicrobiano sintético que atua como um antimetabólito na mesma via de síntese do ácido fólico das sulfonamidas. No entanto, a trimetoprima é um análogo estrutural do ácido di-hidrofólico e inibe uma etapa posterior na via metabólica (Figura 3) (LOSEE L. et al., 2015).
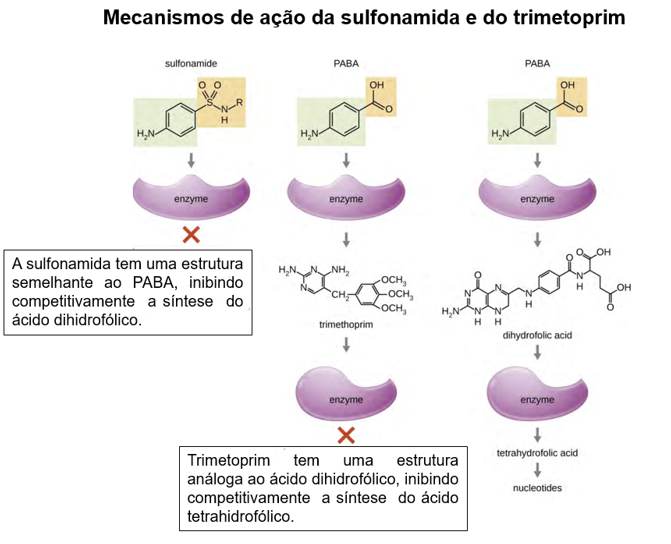
Figura 03: Sulfonamidas e trimetoprima são exemplos de antimetabólitos que interferem na síntese de ácido fólico pelas bactérias, bloqueando a biossíntese de purinas e pirimidinas, inibindo assim o crescimento bacteriano (Adaptado de libretexts.org – Acesso em 01/11/2023 – Diponível em https://query.libretexts.org/Idioma_Portugues/Microbiologia_%28OpenStax%29/14%3A_Medicamentos_antimicrobianos/14.03%3A_Medicamentos_direcionados_a_outros_microrganismos).
O medicamento isoniazida é um antimetabólito com toxicidade específica para micobactérias e tem sido usado há muito tempo em combinação com rifampicina ou estreptomicina no tratamento da tuberculose. Ele é administrado como um pró-fármaco, requerendo ativação por meio da ação de uma enzima intracelular bacteriana peroxidase, formando isoniazida-nicotinamida adenina dinucleotídeo (NAD) e isoniazida-nicotinamida adenina dinucleotídeo fosfato (NADP), o que acaba impedindo a síntese de ácido micólico, essencial para a parede celular das micobactérias. Possíveis efeitos colaterais do uso de isoniazida incluem hepatotoxicidade, neurotoxicidade e toxicidade hematológica (anemia) (PAN J.-X. et al., 2012).
4.6 Inibidor de ATP sintase
A bedaquilina, representando a classe de compostos antibacterianos sintéticos chamada de diarilquinolonas, utiliza um novo modo de ação que inibe especificamente o crescimento das micobactérias. Embora o mecanismo específico ainda não tenha sido elucidado, esse composto parece interferir na função das ATP sintases, possivelmente interferindo no uso do gradiente de íons de hidrogênio para a síntese de ATP por fosforilação oxidativa, levando a uma redução na produção de ATP. Devido aos seus efeitos colaterais, incluindo hepatotoxicidade e potencial arritmia cardíaca letal, seu uso é reservado para casos graves de tuberculose que não podem ser tratados de outra forma (FURUNO J.P. et al., 2014).
4.7 Mecanismos de outros medicamentos antimicrobianos
4.7.1 Medicamentos antifúngicos
Os imidazóis são fungicidas sintéticos que interferem na biossíntese do ergosterol; eles são comumente usados em aplicações médicas e também na agricultura para evitar o mofo em sementes e colheitas. Exemplos incluem miconazol, cetoconazol e clotrimazol, que são usados no tratamento de infecções fúngicas da pele, como o pé de atleta (tinea pedis), a micose da virilha (tinea cruris) e a micose do corpo (tinea corporis). Essas infecções são frequentemente causadas por dermatófitos dos gêneros Trichophyton, Epidermophyton e Microsporum. O miconazol também é usado predominantemente no tratamento de infecções vaginais por leveduras causadas pelo fungo Candida, e o cetoconazol é usado para tratar pitiríase versicolor e caspa, que podem ser causados pelo fungo Malassezia (BERDY J., 2005).
Os medicamentos triazólicos, incluindo o fluconazol, também inibem a biossíntese do ergosterol. No entanto, podem ser administrados por via oral ou intravenosa para o tratamento de vários tipos de infecções sistêmicas por leveduras, incluindo candidíase oral e meningite criptocócica, ambas prevalentes em pacientes com AIDS. Os triazóis também exibem uma toxicidade mais seletiva em comparação com os imidazóis e estão associados a menos efeitos colaterais (GUL S. et al., 2019)
As aminas alílicas, uma classe de medicamentos antifúngicos sintéticos com estrutura diferente, inibem uma etapa anterior na biossíntese do ergosterol. A amina alílica mais comumente usada é a terbinafina (comercializada sob o nome Lamisil), que é usada topicamente no tratamento de infecções fúngicas da pele causadas por dermatófitos, como o pé de atleta, a micose e a micose da virilha. O tratamento oral com terbinafina também é usado para tratar fungos nas unhas das mãos e dos pés, mas pode estar associado ao raro efeito colateral de hepatotoxicidade (FANORO, O.T.; PARANI, S.; MALULEKE, R.; et al., 2021).
Os polienos são uma classe de agentes antifúngicos naturalmente produzidos por certas bactérias do solo actinomiceto e estão relacionados estruturalmente aos macrolídeos. Essas grandes moléculas lipofílicas se ligam ao ergosterol nas membranas citoplasmáticas dos fungos, criando poros. Exemplos comuns incluem nistatina e anfotericina B. A nistatina é tipicamente usada como tratamento tópico para infecções por leveduras na pele, boca e vagina, mas também pode ser usada para infecções fúngicas intestinais. O medicamento anfotericina B é usado para infecções fúngicas sistêmicas, como aspergilose, meningite criptocócica, histoplasmose, blastomicose e candidíase. A anfotericina B foi o único medicamento antifúngico disponível por várias décadas, mas seu uso está associado a alguns efeitos colaterais graves, incluindo nefrotoxicidade (toxicidade renal) (LOWY F.D., 2003).
4.7.2 Drogas antiprotozoários
Existem vários mecanismos pelos quais os medicamentos antiprotozoários atacam protozoários infecciosos. Alguns são antimetabólitos, como a atovaquona, o proguanil e as artemisininas. A atovaquona, além de ser antifúngica, bloqueia o transporte de elétrons em protozoários e é usada no tratamento de infecções por protozoários, incluindo malária, babesiose e toxoplasmose. O proguanil é outro antimetabólito sintético que é processado nas células parasitárias em sua forma ativa, inibindo a síntese de ácido fólico em protozoários. Ele é frequentemente usado em combinação com a atovaquona, e a combinação é comercializada como Malarone tanto para o tratamento quanto para a prevenção da malária (IMPERI F. et al., 2013).
A artemisinina, um antifúngico de origem vegetal descoberto pela primeira vez por cientistas chineses na década de 1970, é bastante eficaz contra a malária. Derivados semissintéticos da artemisinina são mais solúveis em água do que a versão natural, o que os torna mais biodisponíveis. Embora o mecanismo exato de ação não seja claro, as artemisininas parecem atuar como pró-fármacos que são metabolizados pelas células-alvo para produzir espécies reativas de oxigênio (ROS) que danificam as células-alvo. Devido ao aumento da resistência aos medicamentos antimaláricos, as artemisininas também são comumente usadas em combinação com outros compostos antimaláricos na terapia baseada em combinações de artemisinina (ACT) (COSTA, J.P. et al., 2021).
Um outro tipo de medicamento antiprotozoário sintético que por muito tempo se acreditou interferir especificamente na replicação de DNA em certos patógenos é a pentamidina. Historicamente, ela tem sido usada para o tratamento da doença do sono africana (causada pelo protozoário Trypanosoma brucei) e leishmaniose (causada por protozoários do gênero Leishmania), mas também é um tratamento alternativo para o fungo Pneumocystis. Alguns estudos indicam que ela se liga especificamente ao DNA encontrado nos cinetoplastos (kDNA; estruturas semelhantes a mitocôndrias presentes apenas em tripanossomas), levando à clivagem do kDNA. No entanto, o DNA nuclear tanto do parasita quanto do hospedeiro permanece inalterado. A pentamidina também parece se ligar ao tRNA, inibindo a adição de aminoácidos ao tRNA, o que impede a síntese de proteínas. Possíveis efeitos colaterais do uso de pentamidina incluem disfunção pancreática e danos no fígado (PEARLMAN B.L., 2012).
As quinolinas são uma classe de compostos sintéticos relacionados à quinina, que tem uma longa história de uso contra a malária. Acredita-se que as quinolinas interfiram na desintoxicação do heme, que é necessária para a quebra eficaz da hemoglobina em aminoácidos pelo parasita dentro das células vermelhas do sangue. Os derivados sintéticos, como cloroquina, quinacrina (também chamada de mepacrina) e mefloquina, são comumente usados como antimaláricos, e a cloroquina também é usada para tratar amebíase, geralmente causada pela Entamoeba histolytica. O uso profilático a longo prazo de cloroquina ou mefloquina pode resultar em efeitos colaterais graves, incluindo alucinações ou problemas cardíacos. Pacientes com deficiência de glicose-6-fosfato desidrogenase sofrem de anemia grave quando tratados com cloroquina (KALOKHE A.S. et al., 2013).
4.7.3 Medicamentos anti-helmínticos
Devido ao fato de os helmintos serem eucariontes multicelulares, assim como os seres humanos, desenvolver medicamentos com toxicidade seletiva contra eles é extremamente desafiador. Apesar disso, várias classes eficazes foram desenvolvidas. Os benzimidazóis sintéticos, como mebendazol e albendazol, se ligam à β-tubulina helmíntica, impedindo a formação de microtúbulos. Os microtúbulos nas células intestinais dos vermes parecem ser particularmente afetados, levando a uma redução na captação de glicose. Além de sua atividade contra uma ampla variedade de helmintos, os benzimidazóis também são ativos contra muitos protozoários, fungos e vírus, e seu uso para inibir a mitose e a progressão do ciclo celular em células cancerígenas está sendo estudado. Possíveis efeitos colaterais de seu uso incluem danos no fígado e supressão da medula óssea (JURGEIT A. et al., 2012).
Os avermectinas são membros da família dos macrolídeos que foram descobertos pela primeira vez a partir de um isolado de solo japonês, Streptomyces avermectinius. Um derivado semissintético mais potente da avermectina é a ivermectina, que se liga aos canais de cloro sensíveis ao glutamato específicos de invertebrados, incluindo helmintos, bloqueando a transmissão neuronal e causando fome, paralisia e morte dos vermes (CALDEIRÃO, A.C.M. et al. 2021).
4.7.4 Medicamentos antivirais
Diferentemente da estrutura complexa de fungos, protozoários e helmintos, a estrutura viral é simples, consistindo de ácido nucleico, uma capa de proteína, enzimas virais e, às vezes, um envelope lipídico. Além disso, os vírus são patógenos intracelulares obrigatórios que utilizam a maquinaria celular do hospedeiro para se replicar. Essas características tornam difícil o desenvolvimento de medicamentos com toxicidade seletiva contra vírus (FONG D.H., BERGHUIS A.M., 2002).
Muitos medicamentos antivirais são análogos de nucleosídeos e funcionam inibindo a biossíntese do ácido nucleico. Por exemplo, o aciclovir (comercializado como Zovirax) é um análogo sintético do nucleosídeo guanosina. Ele é ativado pela enzima viral do herpes simples, a timidina quinase, e, quando adicionado a uma cadeia de DNA em crescimento durante a replicação, causa a terminação da cadeia. Sua especificidade para células infectadas por vírus decorre da necessidade de uma enzima viral para ativá-lo e da maior afinidade da forma ativada com a DNA polimerase viral em comparação com a DNA polimerase da célula hospedeira (IMPERI F. et al., 2013).
O aciclovir e seus derivados são frequentemente usados no tratamento de infecções por vírus do herpes, incluindo herpes genital, catapora, herpes-zóster, infecções pelo vírus Epstein-Barr e infecções pelo citomegalovírus. O aciclovir pode ser administrado topicamente ou sistemicamente, dependendo da infecção. Um possível efeito colateral de seu uso inclui a nefrotoxicidade. O medicamento adenina-arabinosídeo, comercializado como vidarabina, é um análogo sintético da deoxiadenosina que possui um mecanismo de ação semelhante ao do aciclovir. Ele também é eficaz no tratamento de vários vírus do herpes humano. No entanto, devido a possíveis efeitos colaterais envolvendo contagens baixas de glóbulos brancos e neurotoxicidade, o tratamento com aciclovir é agora preferido (LOWY F.D., 2003).
A inibição da síntese de ácido nucleico não é o único alvo dos antivirais sintéticos. Embora o modo de ação da amantadina e do ser correlato, a rimantadina, não seja totalmente claro, esses medicamentos parecem se ligar a uma proteína transmembrana que está envolvida na liberação do vírus da influenza dos endossomos. Bloquear a liberação do vírus também impede a liberação de RNA viral nas células hospedeiras e subsequente replicação viral (FANORO O. et al., 2021).
A crescente resistência limitou o uso da amantadina e da rimantadina no tratamento da influenza A. O uso de amantadina pode resultar em efeitos colaterais neurológicos, mas os efeitos colaterais da rimantadina parecem ser menos graves. Curiosamente, devido aos seus efeitos sobre substâncias químicas cerebrais, como a dopamina e o NMDA (N-metil D-aspartato), a amantadina e a rimantadina também são usadas para o tratamento da doença de Parkinson (GUL S., et al., 2019).
4.8 Resistência a medicamentos
A resistência antimicrobiana não é um fenômeno novo. Na natureza, os micróbios estão constantemente evoluindo para superar os compostos antimicrobianos produzidos por outros microrganismos. O desenvolvimento humano de medicamentos antimicrobianos e seu amplo uso clínico simplesmente forneceram mais uma pressão seletiva que promove a evolução contínua. Vários fatores importantes podem acelerar a evolução da resistência a medicamentos. Isso inclui o uso excessivo e inadequado de antimicrobianos, uso inadequado de antimicrobianos, doses subterapêuticas e não conformidade do paciente com o curso de tratamento recomendado (FURUNO J.P. et al., 2014).
4.8.1 Mecanismos de resistência a medicamentos
Existem vários mecanismos comuns de resistência a medicamentos, que são resumidos na Figura 4. Esses mecanismos incluem a modificação enzimática do medicamento, a modificação do alvo antimicrobiano e a prevenção da penetração ou acúmulo do medicamento (KABEERDASS N. et al., 2021).
Figura 04: Existem várias estratégias que os micróbios utilizam para desenvolver resistência a medicamentos antimicrobianos. (Não mostrado: superprodução do alvo, mimetismo do alvo e desvio enzimático). (Crédito: modificação do trabalho de Gerard D. Wright).
4.8.1.1 Modificação do alvo ou inativação da droga
A modificação ou inativação do medicamento ocorre quando genes de resistência codificam enzimas que modificam quimicamente um antimicrobiano, inativando-o, ou destroem um antimicrobiano por hidrólise. A resistência a muitos tipos de antimicrobianos ocorre por meio desse mecanismo. Por exemplo, a resistência aos aminoglicosídeos pode ocorrer por meio da transferência enzimática de grupos químicos para a molécula do medicamento, prejudicando a ligação do medicamento ao seu alvo bacteriano (LOSEE L. et al., 2015; PINHEIRO M. et al., 2021).
Para os β-lactâmicos, a resistência bacteriana pode envolver a hidrólise enzimática da ligação β-lactâmica dentro do anel β-lactâmico da molécula do medicamento. Uma vez que a ligação β-lactâmica é quebrada, o medicamento perde sua atividade antibacteriana. Esse mecanismo de resistência é mediado pelas β-lactamases, que são o mecanismo mais comum de resistência aos β-lactâmicos. A inativação da rifampicina ocorre comumente por meio de glicosilação, fosforilação ou adenosina difosfato (ADP) ribosilação, e a resistência aos macrolídeos e lincosamidas também pode ocorrer devido à inativação enzimática do medicamento ou modificação (NELSON M.L. et al., 2010; SHATAN, A.B. et al., 2021).
4.8.1.2 Superprodução do alvo ou desvio enzimático
Quando um medicamento antimicrobiano atua como um antimetabólito, direcionando-se a uma enzima específica para inibir sua atividade, existem maneiras adicionais pelas quais a resistência microbiana pode ocorrer (THE WHITE HOUSE, 2015).
Primeiro, o microrganismo pode superproduzir a enzima alvo, de modo que haja uma quantidade suficiente de enzima livre de antimicrobiano para realizar a reação enzimática adequada (TORTELLA, G., 2021).
Segundo, a célula bacteriana pode desenvolver uma via alternativa que contorna a necessidade da enzima alvo funcional. Ambas essas estratégias foram encontradas como mecanismos de resistência aos sulfonamidas. A resistência à vancomicina em S. aureus foi demonstrada envolver a diminuição da ligação cruzada das cadeias peptídicas na parede celular bacteriana, o que proporciona um aumento nos alvos aos quais a vancomicina pode se ligar na parede celular externa. A ligação aumentada de vancomicina na parede celular externa cria um bloqueio que impede que as moléculas de medicamento livre penetrem onde podem bloquear a síntese da nova parede celular (NELSON M.L. et al., 2010).
4.8.1.3 Mimetismo de alvo
Um mecanismo de resistência recentemente descoberto chamado mimetismo de alvo envolve a produção de proteínas que se ligam e sequestram medicamentos, impedindo que os medicamentos se liguem ao seu alvo. Por exemplo, o Mycobacterium tuberculosis produz uma proteína com repetições regulares de pentapeptídeos que parece imitar a estrutura do DNA. Esta proteína se liga às fluoroquinolonas, as sequestrando e impedindo que se liguem ao DNA, proporcionando resistência do M. tuberculosis às fluoroquinolonas. Proteínas que imitam o sítio A do ribossomo bacteriano foram encontradas como contribuintes para a resistência aos aminoglicosídeos também (GUL S. et al., 2019).
4.9 Staphylococcus aureus resistente à meticilina (MRSA)
A meticilina, uma penicilina semi-sintética, foi concebida para resistir à inativação por β-lactamases. Infelizmente, pouco depois de a meticilina ter sido introduzida na prática clínica, surgiram e começaram a espalhar-se estirpes de S. aureus resistentes à meticilina. O mecanismo de resistência, a aquisição de uma nova PBP de baixa afinidade, proporcionou a S. aureus resistência a todos os β-lactâmicos disponíveis (DADGOSTAR P., 2019).
As estirpes de Staphylococcus aureus resistentes à meticilina (MRSA) são agentes patogénicos oportunistas generalizados e constituem uma preocupação especial para as infecções da pele e de outras feridas, mas também podem causar pneumonia e septicemia (FONG D.H., BERGHUIS A.M., 2002).
Embora originalmente constituíssem um problema em contextos de cuidados de saúde (MRSA adquirido em meio hospitalar [HA-MRSA]), as infecções por MRSA são agora também adquiridas através do contato com pessoas contaminadas do público em geral, o chamado MRSA associado à comunidade (CA-MRSA). Aproximadamente um terço da população transporta S. aureus como membro do microbiota nasal normal sem ficar doente, e cerca de 6% destas estirpes são resistentes à meticilina (COSTA, J.P. et al., 2021).
4.10 Enterococos e Staphylococcus aureus resistentes à vancomicina
A vancomicina é eficaz apenas contra organismos gram-positivos e é usada para tratar infecções de feridas, infecções sépticas, endocardite e meningite causadas por patógenos resistentes a outros antibióticos. É considerada uma das últimas linhas de defesa contra infecções resistentes, incluindo o MRSA. Com o aumento da resistência aos antibióticos nas décadas de 1970 e 1980, o uso de vancomicina aumentou, e não é surpreendente que tenhamos visto o surgimento e disseminação de enterococos resistentes à vancomicina (VRE), Staphylococcus aureus resistente à vancomicina (VRSA) e Staphylococcus aureus intermediário à vancomicina (VISA) (FONG D.H., BERGHUIS A.M., 2002).
O mecanismo de resistência à vancomicina entre os enterococos envolve a modificação do alvo, com uma mudança estrutural na parte peptídica dos subunidades do peptidoglicano, impedindo que a vancomicina se ligue. Essas cepas são normalmente transmitidas entre os pacientes em ambientes clínicos por meio do contato com profissionais de saúde e superfícies contaminadas e equipamentos médicos (IMPERI F. et al., 2013).
5 CONSIDERAÇÕES FINAIS
A urgência por novos agentes antimicrobianos em face da crescente resistência microbiana a antibióticos e antifúngicos é uma realidade incontestável, como evidenciado pela declaração da Organização Mundial da Saúde (OMS) e projeções alarmantes para o futuro. A comunidade científica está ativamente engajada em estratégias inovadoras, incluindo a síntese de novos compostos e o estudo de nanopartículas, na busca por soluções eficazes para enfrentar essa crise global de saúde.
Assim, destacamos a relevância da síntese e atividade biológica de agentes antimicrobianos, o que certamente proporcionará uma visão abrangente dos avanços científicos e das pesquisas em andamento. A exploração da evolução da síntese de medicamentos, desde as primeiras drogas antimicrobianas até as mais recentes, oferece uma perspectiva histórica crucial para compreender a complexidade desse campo.
Os fundamentos do uso de antimicrobianos, destacando a produção natural dessas substâncias pela natureza, são essenciais para contextualizar a necessidade de um uso responsável. A discussão sobre medicamentos bacteriostáticos e bactericidas ressalta a importância da escolha adequada com base na infecção e no estado imunológico do paciente.
Em conclusão, este trabalho destaca a importância crítica de avanços contínuos na síntese de agentes antimicrobianos e a compreensão aprofundada de seus mecanismos de ação. A urgência em abordar a resistência microbiana exige esforços colaborativos entre a comunidade científica, profissionais de saúde e formuladores de políticas. O equilíbrio entre inovação e responsabilidade no uso de antimicrobianos é essencial para garantir a eficácia desses medicamentos vitais, preservando-os como ferramentas valiosas no tratamento e controle de doenças infecciosas.
6 REFERÊNCIAS
- ALMOHAYWI, B.; YU, T.T.; ISKANDER, G.; et al., Synthesis of Alkyne-Substituted Dihydropyrrolones as Bacterial Quorum-Sensing Inhibitors of Pseudomonas aeruginosa. Antibiotics 2022, 11, 151. [CrossRef] [PubMed].
- BALTZ M. “Antimicrobials from Actinomycetes: Back to the Future.” Microbe 2 no. 3 (2007):125–131.
- BERDY J. “Bioactive Microbial Metabolites.” The Journal of Antibiotics 58 no. 1 (2005):1–26.
- CALDEIRÃO, A.C.M.; ARAUJO, H.C.; TOMASELLA, C.M.; et al., Effects of Antifungal Carriers Based on Chitosan-Coated Iron Oxide Nanoparticles on Microcosm Biofilms. Antibiotics 2021, 10, 588.
- COSTA, J.P.; SOUSA, S.A.; SOEIRO, C.; et al., Synthesis and Characterization of Camphorimine Au(I) Complexes with a Remarkably High Antibacterial Activity towards B. contaminans and P. aeruginosa. Antibiotics 2021, 10, 1272.
- DADGOSTAR P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [CrossRef] [PubMed].
- DICKINSON B.D. et al. “Drug Interactions between Oral Contraceptives and Antibiotics.” Obstetrics & Gynecology 98, no. 5 (2001):853–860.
- FALAGAS M.E., KARAGEORGOPOULOS D.E. “Adjustment of Dosing of Antimicrobial Agents for Bodyweight in Adults.” The Lancet 375 no. 9710 (2010):248–251.
- FANORO, O.; PARANI, S.; MALULEKE, R.; et al., Room-Temperature Synthesis of Gold Nanoparticles Using Combretum erythrophyllum Leaf Extract: Antibacterial and Cell Viability Studies against Normal and Cancerous Cells. Antibiotics 2021, 10, 893.
- FANORO, O.T.; PARANI, S.; MALULEKE, R.; et al., Biosynthesis of Smaller-Sized Platinum Nanoparticles Using the Leaf Extract of Combretum erythrophyllumand Its Antibacterial Activities. Antibiotics 2021, 10, 1275.
- FONG D.H., BERGHUIS A.M. “Substrate Promiscuity of an Aminoglycoside Antibiotic Resistance Enzyme Via Target Mimicry.” EMBO Journal 21 no. 10 (2002):2323–2331.
- FURUNO J.P. et al. “Using Antibiograms to Improve Antibiotic Prescribing in Skilled Nursing Facilities.” Infection Control and Hospital Epidemiology 35 no. Suppl S3 (2014):S56–61.
- GUL S.; KHAN S.B.; REHMAN I.U.; et al., A Comprehensive Review of Magnetic Nanomaterials Modern Day Theranostics. Front. Mater. 2019, 66, 179. [CrossRef].
- IMPERI F. et al. “New Life for an Old Drug: The Anthelmintic Drug Niclosamide Inhibits Pseudomonas aeruginosa Quorum Sensing.” Antimicrobial Agents and Chemotherapy 57 no. 2 (2013):996-1005.
- JURGEIT A. et al. “Niclosamide Is a Proton Carrier and Targets Acidic Endosomes with Broad Antiviral Effects.” PLoS Pathogens 8 no. 10 (2012):e1002976.
- KABEERDASS, N.; AL OTAIBI, A.; RAJENDRAN, M.; et al., Bacillus-Mediated Silver Nanoparticle Synthesis and Its Antagonistic Activity against Bacterial and Fungal Pathogens. Antibiotics 2021, 10, 1334.
- KALOKHE A.S. et al. “Multidrug-Resistant Tuberculosis Drug Susceptibility and Molecular Diagnostic Testing: A Review of the Literature. American Journal of the Medical Sciences 345 no. 2 (2013):143–148.
- LOSEE L. et al. “A New Antibiotic Kills Pathogens Without Detectable Resistance.” Nature 517 no. 7535 (2015):455–459.
- LOWY F.D. “Antimicrobial Resistance: The Example of Staphylococcus aureus.” Journal of Clinical Investigation 111 no. 9 (2003):1265–1273.
- MARINESCU M. Synthesis of Antimicrobial Benzimidazole–Pyrazole Compounds and Their Biological Activities. Antibiotics 2021, 10, 1002. [CrossRef] [PubMed].
- MATÍAS F. MARTÍNEZ, LUIS A. QUIÑONES. Relationship Between Pharmacokinetics and Pharmacogenomics and Its Impact on Drug Choice and Dose Regimens. ADME Processes in Pharmaceutical Sciences, 2018. ISBN : 978-3-319-99592-2.
- NELSON M.L. et al. “Brief Communication: Mass Spectroscopic Characterization of Tetracycline in the Skeletal Remains of an Ancient Population from Sudanese Nubia 350–550 CE.” American Journal of Physical Anthropology 143 no. 1 (2010):151–154.
- PAN J.-X. et al. “Niclosamide, An Old Antihelminthic Agent, Demonstrates Antitumor Activity by Blocking Multiple Signaling Pathways of Cancer Stem Cells.” Chinese Journal of Cancer 31 no. 4 (2012):178–184.
- PEARLMAN B.L. “Protease Inhibitors for the Treatment of Chronic Hepatitis C Genotype-1 Infection: The New Standard of Care.” Lancet Infectious Diseases 12 no. 9 (2012):717–728.
- PINHEIRO M.; COSTA J.; MARQUES F.; et al., Bioactive Coatings with Ag-Camphorimine Complexes to Prevent Surface Colonization by the Pathogenic Yeast Candida albicans. Antibiotics 2021, 10, 638. [CrossRef] [PubMed].
- SHATAN, A.B.; PATSULA, V.; DYDOWICZOVÁ, A.; et al., Cationic Polymer-Coated Magnetic Nanoparticles with Antibacterial Properties: Synthesis and In Vitro Characterization. Antibiotics 2021, 10, 1077. [CrossRef] [PubMed].
- THE WHITE HOUSE. National Action Plan for Combating Antibiotic-Resistant Bacteria. Washington, DC: The White House, 2015.
- TORTELLA, G.; RUBILAR, O.; FINCHEIRA, P.; et al., Bactericidal and Virucidal Activities of Biogenic Metal-Based Nanoparticles: Advances and Perspectives. Antibiotics 2021, 10, 783. [CrossRef] [PubMed].
- Treatment of War Wounds: A Historical Review. Clinical Orthopaedics and Related Research 467 no. 8 (2009):2168–2191.
- TREIBER L.; PEZOLT C.; ZENG H.; et al., Dual Agents: Fungal Macrocidins and Synthetic Analogues with Herbicidal and Antibiofilm Activities. Antibiotics 2021, 10, 1022. [CrossRef] [PubMed].
- VAZ, L.E., et al. “Prevalence of Parental Misconceptions About Antibiotic Use.” Pediatrics 136 no.2 (August 2015). DOI: 10.1542/ peds.2015-0883.
- VERMA S., SINGH S.P. “Current and Future Status of Herbal Medicines.” Veterinary World 1 no. 11 (2008):347–350.
- WAINWRIGHT V. “Moulds in Ancient and More Recent Medicine.” Mycologist 3 no. 1 (1989):21–23.
- WILLIAMS, J.D. β-lactamases and β-lactamase inhibitors. Inter. J. Antimicrob. Agents, 12: 3-7, 1999.
1Biomédico pelo Centro Universitário UNA – MG, Mestre em Bioquímica e Imunologia pela Universidade Federal de Minas Gerais – UFMG, Doutorando em Educação pela Universidad Internacional Iberoamericana – Porto Rico (EUA), Especialista em Analises Clinicas e Microbiologia pela Faculdade Venda Nova do Imigrante – ES (FAVENI – ES);
2Biomédica pela Universidade Paulista (UNIP), Aluna do curso de Pós-Graduação Lato Sensu em Hematologia Clínica e Banco de Sangue, Hemoterapia e Terapia Celular pelo Instituto Monte Pascoal – GO;
3Biólogo pela Faculdade Araguaia – GO (UniAraguaia), Aluno do curso de Pós-Graduação Lato Sensu em Microbiologia Aplicada ao Laboratório Clínico pelo Instituto Monte Pascoal – GO;
4Enfermeira pela Faculdade Padrão – GO, Especialista em UTI e Urgência e Emergência pela Faculdade Afirmativo – MT;
5Acadêmica do curso de Nutrição pelo Centro Universitário Leonardo da Vinci (UNIASSELVI);
6Acadêmica do curso de Biomedicina pelo Centro Universitário Leonardo da Vinci (UNIASSELVI); Técnica em Enfermagem pelo Instituto de Técnologia e Educação de Goiás – GO (ITEG);
7Biomédico pela Pontifícia Católica de Goiás – GO (PUC-GO), Mestre em Genética Pontifícia Católica de Goiás – GO (PUC-GO);Especialista em Gestão e Biossegurança em Estética e Cosmética pela Universidade Estadual de Goiás – GO (UEG);
8Biomédica pela Universidade Paulista (UNIP), Aluna do curso de Pós-Graduação Lato Sensu em Hematologia Clínica e Bioinformática pela Faculdade Unyleya – DF;
9Biomédico pela Universidade Paulista (UNIP), Aluno do curso de Pós-Graduação Lato Sensu em Hematologia Clínica e Banco de Sangue, Hemoterapia e Terapia Celular pelo Instituto Monte Pascoal – GO;
10Licenciado em Letras pela Universidade Candido Mendes – RJ, Doutor em Educação pela Universidade São Francisco – SP (USF) Mestre em Educação pela Universidade Estadual de Minas Gerais – MG (UEMG), Especialista em Ensino da Língua Portuguesa pela Universidade Candido Mendes – RJ.
Instituição: 1,2,4,6,7,8Unidade de Pronto Atendimento 24 horas (UPA 24 horas) / Secretaria Municipal de Saúde do Município de Senador Canedo – GO; 2,3,9Instituto Monte Pascoal – GO; 1,5,6Centro Universitário Leonardo da Vinci (UNIASSELVI); 10Universidade Federal de Minas Gerais – MG (UFMG).