REGISTRO DOI: 10.5281/zenodo.10201667
Milene Fernandes Farias¹
Aline Brasil Aranha²
Jaime Giovany Arnez Maldonado²
RESUMO
A síndrome de Andersen-Tawil trata-se de uma doença rara congênita de herança autossômica dominante caracterizada por arritmias ventriculares, paralisia periódica e dimorfismo. É uma condição em que ocorre alteração de canal iônico cardíaco, classificada como tipo 7 da síndrome do QT longo congênito. O relato apresenta uma mulher jovem, 27 anos, sem comorbidades, admitida na emergência, com queixa de fraqueza nos membros inferiores. Inicialmente o médico plantonista suspeitou de um quadro de síndrome de Guillain Barré, entretanto durante a anamnese dirigida, foi relatado a história de episódio semelhante autolimitado há seis anos. Após realização de exames complementares houve a confirmação da síndrome do QT longo 7 através da presença de alterações dismórficas, arritmia ventricular, paralisia flácida e mutação encontrada no gene KCNJ2.
Palavras-chave: Síndrome de Andersen-Tawil; Síndrome do QT Longo, arritmia cardíacas, doenças raras, paralisia.
ABSTRACT
Andersen-Tawil syndrome is a rare congenital disease of autosomal dominant inheritance characterized by ventricular arrhythmias, periodic paralysis and dimorphism. It is a condition in which there is alteration of the cardiac ion canal, classified as type 7 of congenital long QT syndrome. The report presents a young woman, 27 years old, without comorbidities, admitted to the emergency room, complaining of weakness in the lower limbs. Initially the doctor on duty suspected Guillain Barrè’s syndrome, however during the directed anamnesis, the history of a similar self-limited episode six years ago was reported. After complementary tests, long QT syndrome 7 was confirmed through the presence of dysmorphic changes, ventricular arrhythmia, flaccid paralysis and mutation found in the KCNJ2 gene.
Keywords: Andersen-Tawil Syndrome; Long QT Syndrome, arrhythmias Cardiac; rare diseases, paralysis.
1. INTRODUÇÃO
A síndrome de Andersen é uma das paralisias periódicas que associada à ectopia ventricular polimórfica (1) foi descrita pela primeira vez em 1963. Inicialmente nomeada em homenagem a Andersen, que em 1971 relatou caso de um menino apresentando fraqueza muscular intermitente, arritmias ventriculares e dimorfismo, descrevendo então a tríade clássica da síndrome. Houve reconhecimento da contribuição dos critérios diagnósticos pelo Dr. Rabi Tawil, passando assim a ser adotada nomenclatura hoje globalmente utilizada de Síndrome de Andersen-Tawil e sua abreviação em inglês “Andersen-Tawil Syndrome” (ATS) (2).
As manifestações não cardíacas e prolongamento QT leve, associados a alta carga de arritmias ventriculares torna essa condição distinta da síndrome do QT longo mais clássica (SQTL) (1) (3).
A ATS, ou paralisia periódica hipocalêmica com arritmia cardíaca, é uma doença autossômica dominante rara caracterizada por episódios de paralisia, arritmias ventriculares e características dimórficas (baixa estatura, hipertelorismo ocular, clinodactilia, micrognatia, implantação baixa de orelhas) (3). Estudos genéticos subsequentes identificaram mutações em KCNJ2- codificado Kir2.1, canal retificador de potássio, como a causa raiz para alguns casos de ATS que levaram a designação de KCNJ2- doença mediada como síndrome do QT longo tipo 7 (LQTST7) no lócus 17q23-q24.2, gerando a canalopatia dos canais de K mediados pela Proteína G. No entanto, a denominação ATS1, decorrente de tais mutações KCNJ2, é preferível ao LQT7S, uma vez que esses pacientes virtualmente nunca exibem um verdadeiro prolongamento QT, mas em vez disso exibem o prolongamento QTU (Intervalo QT mais a onda U) síndrome do QT longo e que as outras mutações genéticas que cursam com síndrome de ATS são agrupadas como ATS tipo 2 (3).
Os canais Kir2.1 são extremamente importantes para os sistemas musculoesquelético e cardíaco. Quando ocorre a disfunção na corrente fornecida por eles, denominada IK1, haverá retardo da repolarização, prolongando a duração do potencial de ação e desestabilização do potencial de repouso da membrana celular (3). Sob a perspectiva da arritmologia, haverá a formação de ectopias ventriculares por meio de pós-despolarizações precoces (terminal-phase) e prolongamento do intervalo QT. Também podem ocorrer pós-despolarizações tardias resultantes da alteração da bomba de troca de sódio-cálcio, ocorrendo acúmulo de cálcio intracelular, semelhante à intoxicação digitálica, mecanismo da taquicardia ventricular bidirecional (4).
Em estudos realizados previamente, autores (1,2,3,4,5) relataram que 71% dos pacientes com confirmação da mutação genética (KCNJ2) possuíam onda T terminal negativa, junção da onda T-U alargada, ondas U bifásicas e amplas e intervalo QT longo.
Os pacientes afetados pela síndrome de Andersen-Tawil geralmente apresentam na infância crises espontâneas de paralisia, que podem estar associadas a níveis baixos, normais ou elevados de potássio. O fenótipo esquelético e facial característico da síndrome de Andersen-Tawil inclui baixa estatura, hipertelorismo orbitário, ptose palpebral bi ou unilateral, nariz largo, orelhas de implantação baixa, mandíbula hipoplásica, defeitos do palato, clinodactilia, escoliose, hipertireoidismo, atresia vaginal, rim displásico e doença cardíaca estrutural (3)(4).
O tratamento da síndrome de Andersen-Tawil é baseado nos sintomas e não há nenhum estudo terapêutico prospectivo e randomizado encontrado na literatura até a data do dia 18 de fevereiro nas bases do PubMed/MedLine com as palavras chaves deste trabalho. Além disso, a terapêutica é um desafio médico devido aos efeitos opostos da reposição do potássio no intervalo QT e na paresia do músculo esquelético. A elevação da concentração sérica de potássio encurta o intervalo QT e suprime as arritmias ventriculares, mas pode acentuar a fraqueza do músculo esquelético (5).
A síndrome de Andersen-Tawil possui um fenótipo característico, entretanto é subdiagnosticada ou diagnosticada de maneira controversa como um distúrbio estritamente neurológico, em vista da síndrome do neurônio motor inferior (paralisia flácida periférica). Os pacientes com essa condição podem não desenvolver sintomas por um longo tempo, a maioria apresentando-os a partir da segunda década de vida (3).
Quando os sintomas ocorrem, podem ser graves resultando até mesmo em morte súbita. Deste modo, é fundamental aos médicos identificar a doença e compreender sua importância, definir as características e correlacioná-las com as melhores opções terapêuticas (4).
Relatamos o caso de uma mulher, amazonense, natural e procedente de Manaus, 27 anos que apresenta características clínicas de ATS e mutação no gene KCNJ2 identificada através de mapeamento genético, foi acompanhada pelo serviço de Clínica Médica/Cardiologia do Hospital Adventista de Manaus durante internação, quando a patologia foi aventada como hipótese diagnóstica e mantem seguimento ambulatorial periódico no serviço de cardiologia.
2. OBJETIVOS
2.1. Objetivo geral
Contribuir com o conhecimento da sociedade médica através da identificação da doença e compreensão de sua importância, principalmente do ponto de vista do diagnóstico clínico e molecular, com ênfase nas manifestações cardíacas.
2.2. Objetivos específicos
a) Reforçar por meio de um relato de caso a importância das manifestações clínicas e de exames complementares da ATS;
b) Descrever o tratamento instituído e o seguimento clínico preconizados para o caso descrito.
3. METODOLOGIA
O estudo proposto trata-se de um relato de caso, observacional e descritivo, desenvolvido no Hospital Adventista de Manaus, iniciado em setembro do ano de 2021. Os instrumentos utilizados para a coleta de informações foram a anamnese e exame físico no período em que a paciente se manteve internada na referida unidade hospitalar- tanto no centro de terapia intensiva (CTI) quanto no Posto de enfermaria clínico, por meio de revisão de prontuário. Também fizeram parte da aquisição de dados a observação e a discussão do caso nas reuniões multidisciplinares do serviço. A finalização da coleta de informações se deu pela aquisição dos dados secundários com revisão contínua do prontuário eletrônico e registro fotográfico dos métodos diagnósticos ao qual paciente foi submetida e revisão de literatura (até a data do dia 18 de fevereiro do presente ano de 2022 nas bases do PubMed/MedLine com as palavras chaves deste trabalho). Todos os registros e informações obtidas foram devidamente explicadas e solicitadas à paciente, a qual de comum acordo e sem fins lucrativos assinou termo de consentimento livre e esclarecido (TCLE).
4. ASPECTOS ÉTICOS
A abordagem foi realizada por médicos qualificados e com formação ética para a minimização do constrangimento, a confidencialidade, o respeito do sigilo da identidade à em todas as fases da pesquisa, salientando que não há direito a remuneração como benefício extra por autorizar a publicação do caso que será apenas divulgado em revista científica. A paciente concordou com sua participação na pesquisa através da assinatura do Termo de Consentimento Livre e Esclarecido (Anexo A).
5. JUSTIFICATIVA
Há múltiplas controvérsias em relação a avaliação de risco na síndrome de Andersen Tawil, na identificação dos preditores da ocorrência de arritmias graves, no tratamento e na interpretação do ECG. Estudar os riscos de parada cardíaca e morte súbita cardíaca (MSC) é difícil devido à falta de bancos de dados confiáveis e à baixa prevalência da própria ATS (4-8).
Por se tratar de uma síndrome rara com dificuldades para diagnóstico e tratamento clínico efetivo, é de extrema importância a divulgação desta apresentação clínica, as etapas diagnósticas e o tratamento proposto, sendo assim compartilhadas informações acerca da evolução clínica apresentada, contribuindo para discussão de casos semelhantes posteriormente relatados.
6. RELATO DE CASO
Uma mulher de 27 anos de idade deu entrada no serviço de pronto atendimento do Hospital Adventista de Manaus, com relato de história de fraqueza muscular em membros inferiores e quatro quedas da própria altura de início 24h antes da admissão no pronto atendimento. Negou doenças infecciosas e imunização nos 30 dias anteriores ao atendimento. Negou também fraqueza em membros superiores, dispneia, disfonia, cefaleia e febre. Não ocorreu ascendente dos sintomas e disautonomias. Relatou quadro semelhante prévio seis anos antes do novo evento, com história de arritmia (bradicardia). Estava em acompanhamento médico e sem tratamento medicamentoso. Negou uso de medicações sem orientação profissional.
Ao exame físico: regular estado geral, hidratada, corada, hemodinamicamente estável, pupilas isocóricas, reflexo fotomotor direto e consensual preservado, eudiadococinesia, marcha claudicante, força muscular grau 3 (Medical Research Conciul) em extensores e flexores do pé e 4 – (menos) em flexores e extensores da coxa, membros superiores: força muscular grau 5 com amplitude dos movimentos preservados reflexos motores (tricipital, bicipital, estilorradial, patelar e aquileu) e sensibilidade preservados (profunda: cinético postural e superficial: dor e sensibilidade geral). Escala de cincinnati: zero.
Exames complementares admissionais dentro da normalidade (sódio, potássio, hemograma, magnésio, cálcio, TSH, T4L, Ureia, Creatinina, TGO, TGP, Bilirrubinas, séricos e Raio X de tórax) e procedida internação do paciente.
Durante internação manifestou bradicardia registrada no eletrocardiograma (ECG) de 12 derivações, sem repercussão hemodinâmica e assintomática, com frequência cardíaca de 32 bpm, mantendo fraqueza distal de membros. Paciente apresentou dois novos episódios de bradicardia- FC: 27 bpm e 34 bpm, ECG ritmo sinusal com bigeminismo ventricular e intervalo QTc prolongado, 518 ms (Figura 1) corrigido pela fórmula de Bazett.
Procedida investigação com avaliação eletrocardiográfica e estrutural cardíaca com monitorização eletrocardiográfica dinâmica de 24h (Holter 24h), Ecocardiograma Transtorácico e USG (ultrassonografia) com doppler fluxometria de tireóide, os dois últimos sem alterações, assim como a função tireoidiana e eletrólitos (Sódio e Potásio séricos). Ao Holter de 24h apresentou extrassístoles ventriculares (EEVV) com FC média de 66 bpm, EEVV polimórficas com densidade alta. Durante a primeira semana manteve em média cinco episódios de bradicardia assintomáticas. Também foi solicitada sorologia para Chagas com resultado negativo.
Evoluiu com melhora progressiva da paresia espontaneamente e com marcha atípica e normal desde as primeiras 24 horas pós admissão hospitalar, recebendo alta médica pela equipe da neurologia para investigação ambulatorial do quadro neurológico e manteve episódios de bradicardia assintomática sendo acompanhada pela equipe de cardiologia.
O resultados apresentados no teste ergométrico, focos ectópicos, presença de onda U e aumento do QT, foram determinantes para a realização de estudo eletrofisiológico com ablação cardíaca de focos ectópicos.
Paciente foi submetida à ablação de ectopias ventriculares, porém o procedimento não obteve sucesso, em vista retorno das ectopias ventriculares no pós operatório imediato do procedimento. O exame ecocardiográfico transesofágico realizado no intraoperatório não demonstrou derrame pericárdico nem lesões valvares importantes.
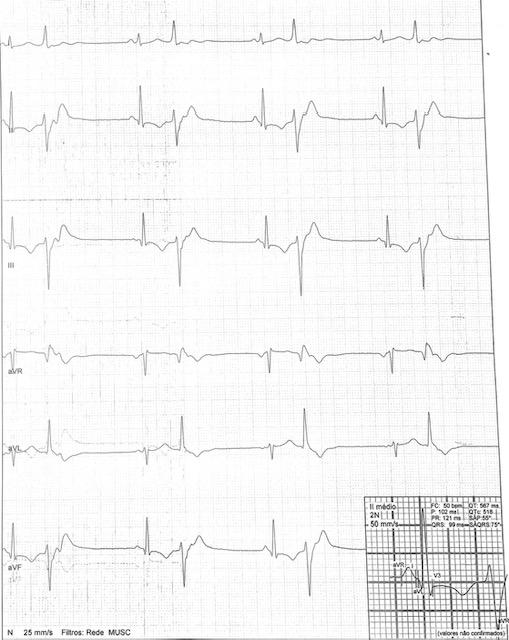
Figura 1 ECG com ritmo sinusal com bigeminismo ventricular e intervalo QTc prolongado.
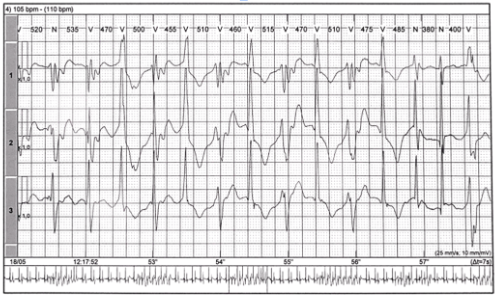
Figura 2 Holter apresentando arritmias ventriculares (35,779, com 2203 episódios de bigeminismo.
Realizou-se ablação, então, no músculo papilar, também sem resposta, havendo reinício do bigeminismo ventricular no pós-operatório.
Paciente recebeu alta com Verapamil e AAS e encaminhada para a avaliação de necessidade de implante de monitor de eventos (Looper implantável), implantação marcapasso (MCP) somente ou MCP com desfibrilador e para avaliação e investigação com geneticista para ATS.
Foi confirmada a mutação no gene KCNJ2 do mapeamento genético, ratificando o diagnóstico da ATS, somados pelas alterações clínicas- extrassístoles ventriculares frequentes, paralisia periódica e as características dimórficas descritas para a síndrome como micrognatia, clinodactilia , hipertelorismo e implantação baixa de orelhas (Figura 3).
Atualmente segue em acompanhamento ambulatorial com arritmologista, em uso de composto de Glicinato de Magnésio e Piridoxina B6 e Diltiazem (bloqueador de canal de cálcio), sem novos episódios de paralisias. Mantem alterações eletrocardiográficas no ECG, bigeminismo com extrassístoles ventriculares, e no Holter presença de extrassístoles ventriculares (39916 episódios) e taquicardia ventricular episódicas. O ecocardiograma atual demonstra redução da fração de ejeção ventricular, no limite inferior da normalidade.
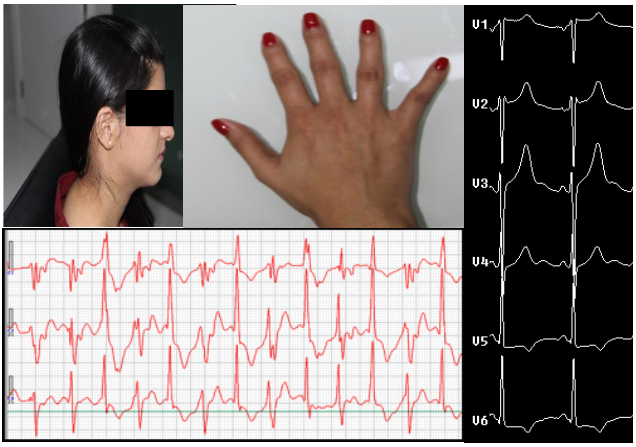
Figura 3 Dimorfismo e alterações cardíacas em portadoras de síndrome de Andersen-Tawil: implantação baixa de orelhas, clinodactilia, micrognatismo e Holter compresença de bigeminismos.
7. DISCUSSÃO
A ATS é um distúrbio autossômico dominante raro e esporádico que abrange alterações fenótipicas e arritmias cardíacas – onda U pronunciada e/ou extrassístoles ventriculares frequentes. Em geral, para o diagnóstico é necessário que o indivíduo apresente pelo menos duas características seguintes: a) Paralisia periódica, b) Pelo menos duas alterações dismórficas (micrognatia, hipertelorismo, clinodactilia, sindactilia, implantação baixa de orelha), c) Mutação KCNJ2 com anormalidades eletrocardiográficas (ondas U alargadas, ectopia ventricular, morte súbita por arritmia ventricular, intervalo QTc aumentado) (1)(6).
Para o diagnóstico definitivo de pacientes que apresentem apenas uma das três alterações descritas é necessário que exista pelo menos um membro da família com diagnóstico estabelecido (Tabela 1) (3). Esses critérios diagnósticos são confirmados e utilizados nos poucos relatos descritos até o momento, e conforme novos casos são estudados, novas características associadas aos portadores da ATS vão sendo incorporadas ao diagnóstico.
Uma das casuísticas confirma o uso dos referidos critérios diagnósticos no caso de paciente ser uma mulher de 20 anos com ATS geneticamente confirmada desde os 12 anos (gene KCJN2), e com características dimórficas típicas da ATS (hipertelorismo, hipoplasia mandibular, clindactilia e sindactilia) somadas a história familiar de pai com paralisia periódica (7). É importante ressaltar que as características dimórficas faciais graves não possuem correlação com o envolvimento cardíaco ou musculoesquelético nos casos descritos até o momento (5).
No presente estudo, relatamos uma paciente com uma mutação KCNJ2 e manifestações clínicas típicas, sem história familiar de morte súbita cardíaca, paralisia periódica ou outra manifestação dismórfica que auxiliassem no diagnóstico de ATS familiar.
A penetrância é altamente variável, assim como a expressão fenotípica e a gravidade da doença manifestadas. A chance de um filho herdar a doença é de 50%, e um dos fatores que podem reduzir essa probabilidade é a não penetrância que ocorre em aproximadamente 6 a 20% dos indivíduos que herdam a mutação, confirmando que as características da síndrome podem ser sutis, o que dificulta o reconhecimento do distúrbio (4) (1) (3).
Caso semelhante divulgado na China em 2015, um menino de 15 anos, mas com história de fraqueza paroxística nos membros desde os dois anos de idade. Foi diagnosticado com ATS, classificado como uma mutação de novo KCNJ2, já que nova mutação heterozigótica não foi encontrada nos genitores (2).
Sendo uma patologia autossômica dominante, os portadores da síndrome terão uma combinação de Kir2.1 normal e mutantes. Os canais mutantes interrompem a ação dos canais normais, causando o que se caracteriza por efeito negativo dominante, o que gera a redução na corrente de potássio (9).
Essa alteração nas propriedades elétricas da membrana celular promovem os surtos de paralisia e arritmias cardíacas. Em alguns pacientes, um fenótipo cardíaco pode ser a única manifestação apresentada. O amplo espectro de anormalidades cardíacas como taquicardia ventricular bidirecional e bigeminismo são as arritmias mais comumente descritas (8).
A paciente do nosso estudo apresentou grande número de EEVV polimórficas (48,62%), bigeminismo e taquicardia ventricular polimórfica não sustentada no holter de 24 horas realizado durante internação. O intervalo QTc de 518 ms foi determinado em ECG de 12 derivações, tratando-se de um QT longo.
Foi demonstrado em uma avalição sistemática de eletrocardiogramas em portadores de ATS, (independentemente da mutação genética) que apenas 17% possuíam intervalo QT > 460 ms (4). Confirmando que a paciente deste estudo apresenta características raras por se tratar de uma síndrome pouco diagnosticada, e dentro do espectro da patologia também demonstra característica pouco frequentes, já que a maioria dos casos apresenta intervalo QTu normal.
No teste ergométrico do caso relatado foi observada manutenção de extrassístoles ventriculares em frequências cardíacas abaixo de 100 bpm, e ritmo sinusal quando submetida a frequências cardíacas maiores. O fenótipo cardíaco dela torna-se interessante entre os casos já descritos em relação a presença de bradicardia, e pelo fato de os registros evidenciarem que na presença de FC mais baixas a paciente manifestou mais episódios extrassístoles ventriculares.
A características da nossa paciente divergem da Taquicardia ventricular polimórfica catecolaminérgica (TVPC), também conhecida como TVPC familiar, doença genética que ocorre na ausência de doença cardíaca estrutural ou síndromes associadas. Na TVPC o exercício normalmente precipita as arritmias. Por outro lado, Na ATS, o exercício não é um gatilho consistente de arritmia e há relatos onde a atividade física suprime a ectopia ventricular na SQTL7 (3).
Entretanto, há uma série de relatos de ATS em que os pacientes foram submetidos a testes de estresse onde foram detectadas arritmias, com agravamento durante o exercício em 71% dos casos(11). Diferindo do caso relatado nesta casuística.
No holter da paciente do relato em questão houve registro de 38.064 episódios de arritmias ventriculares, sendo a mais longa com duração de 2,2 segundos. A curta duração dos episódios pode ser a causa para poucos sintomas ou ausência deles, mesmo diante de ectopias ventriculares complexas.
Em uma revisão bibliográfica, foi afirmado que pacientes com ATS apresentam um aumento na dispersão transmural da repolarização que não altera o intervalo QT. Desse modo, a manifestação de palpitações não ocorre já que os múltiplos eventos são de curta duração. Essas características tornam o fenótipo cardíaco menos grave em comparação com outros tipos de síndrome do QT longo (5).
Entretanto, casos complexos com arritmia cardíaca grave com insuficiência respiratória e outros com evolução para morte súbita também já foram descritos (10). A ocorrência de morte súbita cardíaca é relativamente infrequente na ATS quando comparada com outras SQTL.
Uma coorte com 20 pacientes, um terço apresentou síncope e um paciente apresentou parada cardíaca (11). Em outra série de casos publicada, 41% dos pacientes com ATS tipo 1 (39 de 96, de 33 famílias com 24 mutações diferentes) apresentavam arritmias cardíacas no ECG de base, metade das quais eram taquicardia ventricular não sustentada, tipicamente taquicardia ventricular bidirecional e parada cardíaca foi documentada em três casos e em 12 a história familiar foi positiva para morte súbita (11).
Anormalidades de condução foram encontradas em 23% dos pacientes em outra coorte realizada, variando de bloqueio atrioventricular de primeiro grau, bloqueio de ramo direito, de ramo esquerdo, bifascicular e atrioventricular de 1o grau (11). Esse achado não foi confirmado em nosso estudo.
A paciente deste estudo manifestou episódios de bradicardia importante durante a internação (FC média de 32 bpm), porém sem registros de bloqueios atrioventriculares nos ECGs registrados. Sabe-se que o batimento ventricular muito precoce pode não gerar pulso na oximetria de pulso, pois não houve tempo suficiente para o enchimento ventricular. Não é infrequente que o comportamento bigeminado com acoplamento muito curto seja confundido com episódio de bradicardia.
As características cardíacas da nossa paciente fazem-nos refletir sobre a necessidade de discussão da associação entre síndrome de Andersen-Tawil e QT longo, colocada em discussão nos estudos recentes (4).
A interação entre QTc longo próprio da ATS com utilização de medicamentos que prolonguem o intervalo QT podem ocasionar desfechos desfavoráveis, sendo prudente avaliar a terapêutica a ser utilizada nesses pacientes com intuito de minimizar danos. A paciente relatada negou uso de medicações de uso contínuo, assim como não fez uso de medicamentos para tratamento de sintomas agudos.
Um relato de caso em que paciente alemão, do sexo masculino esquizofrênico, e portador de ATS (histórico de ectopias ventriculares polimórficas e paralisia periódica) fazia uso de amissulprida, uma das medicações que também prolongam intervalo QT (Tabela 2). Essa associação foi responsável para a ocorrência de morte súbita pela sobreposição de fatores congênitos e adquiridos (12).
Ainda não há recomendações definitivas para o manejo das arritmias ventriculares. Não existe grande evidência científica quanto ao tratamento com uso de antiarrítmicos. As terapêuticas utilizadas nos relatos publicados são geralmente direcionadas para os sintomas específicos de cada paciente, ficando a critério do médico assistente a terapêutica a ser instituída. Os dados de eficácia, portanto, são empíricos, conflitantes e heterogêneos (4)(11).
Os objetivos principais da terapêutica com as medicações para controle das arritmias ventriculares (AV) são a diminuição da densidade das ectopias e supressão da taquicardia ventricular. As evidências atuais indicam que a manutenção de um intervalo QTc curto para prevenir a degeneração em torsade de pointes pode ser alcançada com bloqueio β-adrenérgico, geralmente o propanolol. Isso deve ser combinado com evitar drogas, particularmente outros agentes cardíacos, que prolongam o intervalo QT (Tabela 2) (4)(8)(11) .
O tratamento nos casos relatados incluiu betabloqueadores, bloqueadores de canal de cálcio, amiodarona, flecainida e modalidades invasivas, como implante de cardioversor-desfibrilador implantável (CDI) ou ablação (8).
A paciente do estudo apresenta contraindicação ao uso de betabloqueador, por apresentar bradicardia importante, o que torna um viés compara-la a casos semelhantes relatados, em que a combinação mais bem sucedida foi flecainida e betabloqueadores com redução da carga de arritmia em cinco dos seis pacientes. O efeito antiadrenérgico dos betabloqueadores foi estabelecido e, quanto à flecainida, o efeito de bloqueio dos canais de sódio que inibe a capacidade das despolarizações tardias de gerar movimentos ascendentes completos também foi crucial. (11).
A ablação de batimentos ectópicos, que seriam o gatilho das taquicardias ventriculares sustentadas, foi realizada em outras canalopatias já estudadas. Pesquisadores descreveram que a origem desses batimentos na ATS seria em diferentes partes da rede de Purkinje esquerda. Essa estratégia poderia ser indicada em pacientes com tempestade elétrica, apesar da ausência de relatos de literatura para a síndrome estudada (4).
A paciente do relato foi submetida a realização de estudo eletrofisiológico cardíaco guiado por eco transesofágico e pacemapping para tentativa de ablação de focos ectópicos de estimulação ventricular no músculo papilar posterior e na região basal. O procedimento não foi efetivo, com retorno das ectopias ventriculares imediatamente. É válido ressaltar que o estudo eletrofisiológico não é considerado um potencial preditor na estratificação de risco (4). E que as tentativas de ablação falham em muitos casos, assim como no caso descrito nesta casuística (8).
Em alguns casos de ATS, são indicados implantes de CDI. Em um grupo de 15 pacientes com ATS geneticamente confirmados, um CDI foi implantado em seis casos devido a síncope recorrente (quatro pacientes) ou mesmo parada cardíaca (dois casos) (8).
A paciente do nosso estudo não foi submetida a implante de CDI até o momento por não estar classificada nos critérios de gravidade preconizados de acordo com as diretrizes atuais. O CDI deve ser implantado especialmente em paciente com AV grave resistente à terapia medicamentosa, com síncope, disfunção ventricular esquerda ou após parada cardíaca (8).
O uso de CDI deve ser indicado com cautela, já que a taquicardia ventricular bidirecional típica da ATS é geralmente autoterminante, a síncope na ATS se repete apenas raramente e a morte súbita na ATS também é um evento raro. Vale ressaltar que o implante de CDI em ATS não elimina a AV frequente. Diante dessas evidências, há dificuldade para programação do dispositivo no ATS (4)(8).
A carga de arritmia apresentada por nossa paciente não melhorou consistentemente com nenhuma das combinações utilizadas, entretanto, ela permanece assintomática, sem novos episódios de paralisia e nenhum evento de síncope. Até o momento, a paciente é a única a manifestar características da ATS na família, sem eventos de morte súbita e outras complicações cardíacas nos parentes conhecidos.
Seria interessante fazer avaliação clínica utilizando os critérios diagnósticos e acompanhamento dos familiares da paciente, levando em consideração o fato de estudos relatando pacientes que não apresentavam sintoma cardíaco nem anormalidade no ECG e que foram diagnosticadas com STA apenas pelas alteraçoes dimorficas. No entanto, a variabilidade fenotípica na ATS pode obscurecer o diagnóstico na ausência de história familiar de todas as três características cardinais (2).
O sexo também pode estar relacionado a distintas manifestações. Em uma família, com indivíduos portadores de mutação no gene KCNJ2, prevaleceu as arritmias ventriculares no sexo feminino com manifestações extracardíacas. Enquanto no sexo masculino a paralisia periódica foi mais prevalente (11). Há evidências de que a modulação hormonal da gestação produza um efeito protetor, foi observada redução de ectopia ventricular durante a gravidez na portadora de ATS (4)(5). Uma possibilidade é que alterações hemodinâmicas ou hormonais relacionadas à gestação possam ter efeitos favoráveis na repolarização cardíaca (5).
Por ser uma afecção de acometimento raro, não existe literatura que correlacione algum genótipo ou característica clínica para o desenvolvimento de morte súbita. A paciente relatada nega uso de terapia hormonal oral ou injetável, é nulípara e apresenta ciclo menstrual regular. Nega alterações vaginais, e não possui histórico nos registros da unidade hospitalar de acompanhamento com profissional ginecologista.
Dada a escassez de evidências referentes à assistência ginecológica e correlação de eventos de paralisia, piora ou melhora das alterações cardíacas durante a gestação, parto, puerpério e menopausa de pacientes femininas com ATS é necessária uma abordagem multidisciplinar cautelosa e longitudinal da paciente em evidência a fim de enriquecer nosso banco de dados sobre as evidências de ATS em mulheres e fornecer dados comparativos para futuros estudos (5).
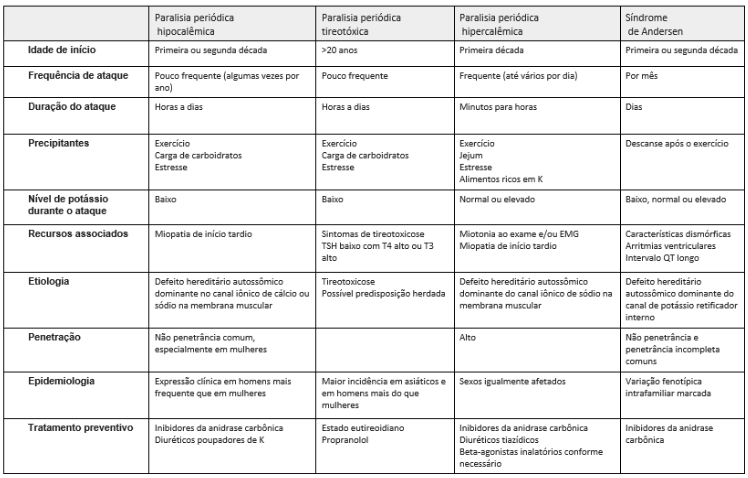
Dentre as paralisias periódicas (Tabela 3), as paralisias periódicas primárias são tradicionalmente classificadas como hiper ou hipocalêmicas, dependendo do nível de potássio sérico durante um ataque. A ATS por sua vez pode se manifestar sem uma mudança hidroeletrolítica, como no caso da paciente deste estudo que estava normocalêmica (13).
Casos com hipocalemia são mais associados a paralisia, que geralmente dura de horas a dias e raramente semanas, incluem repouso após exercício, repouso prolongado e refeições ricas em carboidratos. A ingestão de potássio pode ajudar na resolução do ataque. A frequência e a gravidade dos ataques geralmente diminuem com a idade. Entre os episódios de paralisia, a força muscular é normal, embora uma miopatia fixa possa se desenvolver com a manutenção da periodicidade dos eventos (9).
A paciente do estudo em questão teve acompanhamento nutricional durante o período de internação hospitalar, estava com baixo peso, 44 Kg, Estatura 1,55 m e IMC 18 kg/m2. Manifestava hiporexia, padrão alimentar inapropriado, com pouca ingesta de alimentos saudáveis no cotidiano. No período em que esteve internada, apresentou boa aceitação alimentar. A avaliação do prontuário de seguimento ambulatorial, revelou que ela mantém acompanhamento com endocrinologista e nos registros há relatos sobre a prática de atividade física regular, e preferencialmente no período noturno.
Além disso, relata história de hipotireoidismo na infância, sem comprovação com exames da época, porém mantem níveis de hormônios tireoidianos normais sem necessidade de terapia medicamentosa. A paciente estudada apresenta episódios de piora da astenia, fadiga e cansaço, sem correlação com paralisia. Tem insônia, com 4h sono à noite, associados a período de stress e ansiedade. O último evento de paralisia que motivou nossa pesquisa ocorreu no período noturno. Caso semelhante foi relatado na Índia, onde os episódios de paralisia foram associados à privação de sono e foram aliviados em cerca de 48 h com medicamentos sintomáticos simples (14).
A paciente apresentou melhora significativa da paralisia após a introdução da terapia medicamentosa com espironolactona, reposição de potássio, que foi introduzido para diminuição do intervalo QTu e terapia com bloqueador de canal de cálcio durante a internação hospitalar. O objetivo do tratamento visa reduzir a frequência e a gravidade dos ataques de paralisia e prevenir o desenvolvimento de arritmias ventriculares malignas, ou seja, é um desafio pois a ATS acomete tecidos excitáveis distintos, cada um com características eletrofisiológicas próprias (13) (8).
Para ataques de fraqueza muscular, os pilares farmacológicos visam minimizar a frequência, gravidade e duração dos episódios de paralisia, reduzindo os prejuízos dos episódios na rotina e funções diárias do paciente, como dependência de terceiros para o autocuidado, internações hospitalares, afastamento do trabalho; e evitar que as paralisias periódicas acarretem em fraqueza muscular fixa persistente. Os inibidores da anidrase carbônica (IAC), acetazolamida e diclorfenamida são utilizados com sucesso em outras formas de paralisia periódica, também se mostraram eficazes na ATS (4)(8)(14)(15).
Pacientes com contra-indicações ao uso de IACs podem ter benefício com uso de poupadores de potássio, espironolactona por exemplo, já que pacientes com ATS precisam manter um nível adequado de potássio, corroborando indiretamente para minimizar os surtos de ataques paralíticos e prevenir o prolongamento do intervalo QT, que também pode ser conseguido com a suplementação oral de potássio com formulações de liberação lenta. Além disso, os pacientes devem evitar quaisquer gatilhos conhecidos de seus ataques (14).
Por se tratar de uma síndrome rara não há consenso ou ensaios randomizados sobre terapêuticas preconizadas para o tratamento dos sintomas na ATS. Uma alternativa seria o ensaio com número limitado de pacientes, randomizado e controlado por placebos em paciente únicos, técnica estabelecida e aceita em casos de patologias raras e em estudos pilotos de novas medicações (14). Em conclusão, identificamos uma paciente amazonense típica para ATS com nova mutação de novo no gene KCNJ2 e expandimos ainda mais nosso conhecimento do fenótipo, diagnóstico e tratamento da ATS.
8. CONCLUSÃO
A síndrome raramente foi relatada no Brasil e a importância de uma doença tão rara como a ATS requer estudos e publicações de casos semelhantes a fim de identificar os fatores envolvidos na penetrancia do gene KCNJ2 e criar um banco de dados para correlações dos casos e os tratamentos instituídos.
A ATS é uma canalopatia muscular única porque afeta diferentes membranas excitáveis, cardíacos, musculoesquelético. A medicina molecular evoluiu consideravelmente nossa compreensão desses distúrbios; no entanto, ainda há muito estudo a ser feito para melhor definir as características clínicas e fisiopatológicas e estabelecer tratamentos mais eficazes e reconhecer a gravidade da doença e o padrão autossômico dominante de transmissão.
Os múltiplos sistemas acometidos são responsáveis pelas manifestações complexas da síndrome. O tratamento adequado da ATS deve ser direcionado para tratar os sintomas cardíacos e musculares esqueléticos. Os atuais são baseados em tratamentos eficazes conhecidos nas demais paralisias periódicas e síndromes LQT, que podem não ser ideais devido à fisiopatologia única da ATS, onde os indivíduos frequentemente se mantem assintomáticos, e na maioria dos casos, sem grandes repercussões ou desfechos desfavoráveis.
A terapia farmacológica ideal necessita ser validada. Os betabloqueadores, bloqueadores dos canais de sódio são bons candidatos nos relatos de casos publicados até o momento. A grande variedade de combinações usadas até hoje claramente exige um registro grande, provavelmente mundial, para fornecer respostas quanto aos pacientes elegíveis ao tratamento, e como tratá-los de maneira adequada, sem intervenções desnecessárias ou pouco efetivas.
O risco de uma morte súbita cardíaca é uma ameaça presente para os pacientes com ATS e diante dessa perspectiva, as diferentes manifestações eletrocardiográficas desse distúrbio devem ser reconhecidas e estudadas. A gravidade da doença e o padrão autossômico dominante de transmissão são de importância significativa para o paciente e sua família, sendo necessário estudo longitudinal do caso relatado e dos familiares para reconhecer fatores determinantes na piora ou controle dos sintomas e correlacioná-los aos eventuais surtos de paralisia e manifestações cardíacas por exemplo.
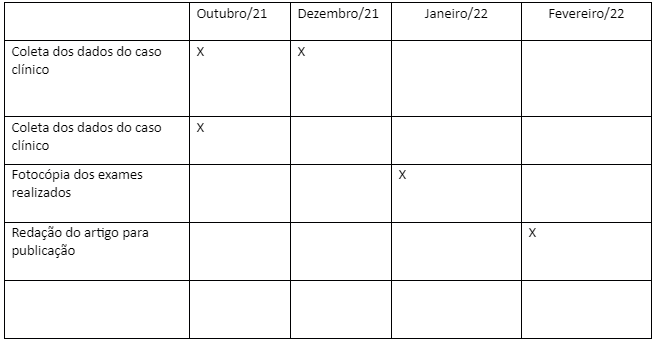
9. CRONOGRAMA
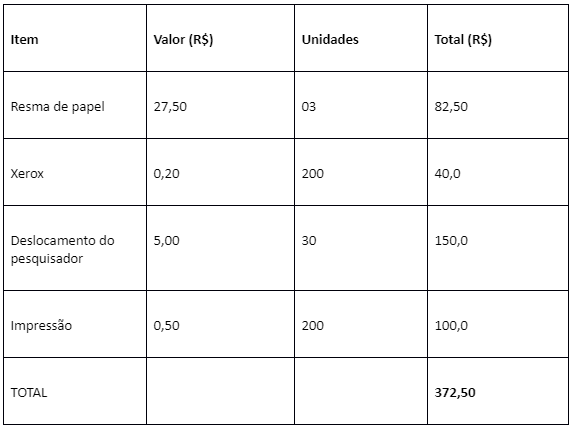
10. ORÇAMENTO
12. ANEXOS
TABELA 1: CRITÉRIOS DIAGNÓSTICOS NA ATS
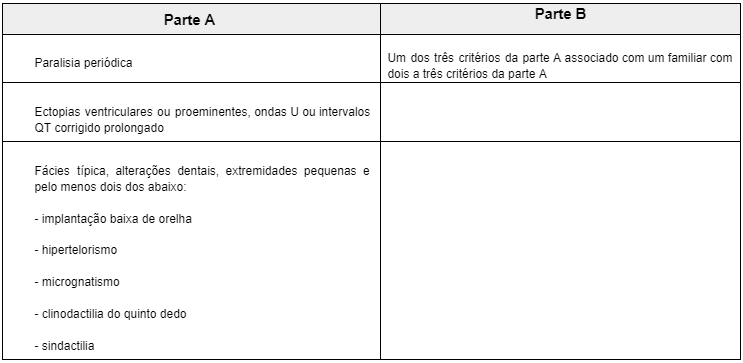
TABELA 2: LISTA DE MEDICAMENTOS QUE CAUSAM PROLOGAMENTO DE QT


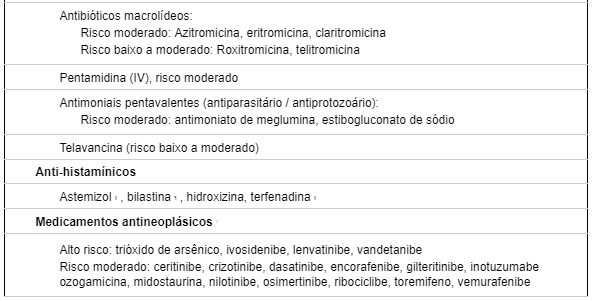
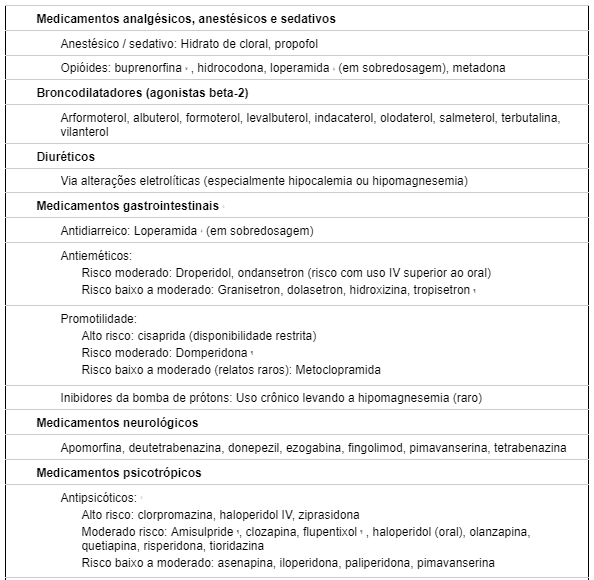

TABELA 3: CARACTERÍSTICAS CLÍNICAS DAS PARALISIAS PERIÓDICAS- DIAGNÓSTICO DIFERENCIAL
9. REFERÊNCIAS BIBLIOGRÁFICAS
1. Almuqbil M, Srour M. Child Neurology: Andersen-Tawil syndrome. Neurology. 17 de março de 2015;84(11):e78–80.
2. Liu X, Huang X, Luan X, Zhou H, Wang T, Wang J, et al. Case report: A Chinese child with Andersen–Tawil syndrome due to a de novo KCNJ2 mutation. J Neurol Sci. maio de 2015;352(1–2):105–6.
3. Tristani-Firouzi M, Etheridge SP. Kir 2.1 channelopathies: the Andersen–Tawil syndrome. Pflüg Arch – Eur J Physiol. julho de 2010;460(2):289–94.
4. Ronsoni R de M, Silvestrini TL. Síndrome de Andersen-Tawil: Síndrome de Andersen-Tawil. J Card Arrhythm. 17 de outubro de 2015;28(3):113–7.
5. Subbiah RN, Gula LJ, Skanes AC, Krahn AD. Andersen-Tawil Syndrome: Management Challenges During Pregnancy, Labor, and Delivery. J Cardiovasc Electrophysiol. setembro de 2008;19(9):987–9.
6. Canon S, Perez N, Beirana LG. Andersen syndrome autosomal dominant in three generations. Am J Med Genet. 16 de julho de 1999;85(2):147–56.
7. Sung RJ, Wu S-N, Wu J-S, Chang H-D, Luo C-H. Electrophysiological mechanisms of ventricular arrhythmias in relation to Andersen-Tawil syndrome under conditions of reduced I K1 : a simulation study. Am J Physiol-Heart Circ Physiol. dezembro de 2006;291(6):H2597–605.
8. Bienias P, Kostera-Pruszczyk A, Bieganowska K, Miszczak-Knecht M, Pruszczyk P. Should a cardioverter-defibrillator be implanted in an Andersen-Tawil syndrome patient with severe ventricular arrhythmias and syncope? :1.
9. Rajakulendran S, Tan SV, Hanna MG. Muscle weakness, palpitations and a small chin: the Andersen-Tawil syndrome. Pract Neurol. 20 de julho de 2010;10(4):227–31.
10. Pyo JY, Joh DH, Park JS, Lee S-J, Lee H, Kim W, et al. Ventricular Tachyarrhythmias in a Patient with Andersen-Tawil Syndrome. Korean Circ J. 2013;43(1):62.
11. Wilde AAM. Andersen-Tawil syndrome, scarier for the doctor than for the patient? Who, when, and how to treat. Europace. 1o de dezembro de 2013;15(12):1690–2.
12. Peters S, Schulze-Bahr E, Etheridge SP, Tristani-Firouzi M. Sudden cardiac death in Andersen–Tawil syndrome. EP Eur. 1o de março de 2007;9(3):162–6.
13. Schoonderwoerd BA, Wiesfeld ACP, Wilde AAM, van den Heuvel F, Van Tintelen JP, van den Berg MP, et al. A family with Andersen-Tawil syndrome and dilated cardiomyopathy. Heart Rhythm. novembro de 2006;3(11):1346–50.
14. Sansone V, Tawil R. Management and treatment of Andersen-Tawil syndrome (ATS). Neurotherapeutics. abril de 2007;4(2):233–7.
15. Modoni A, Bianchi MLE, Vitulano N, Pagliarani S, Perna F, Sanna T, et al. Lack of Any Cardiac Involvement in a Patient with Andersen-Tawil Syndrome Associated with the c.574A→G Mutation in KCNJ2. Cardiology. 2011;120(4):200–3.
16. P JR, P Y, N. V. S, U. V, Elahi SM, Amalakanti S, et al. Vanishing Weakness and Persistent Cardiac Dysrhythmia: Are We Dealing with Andersen Tawil Syndrome? Indian J Pediatr. julho de 2015;82(7):642–4.
17. Márquez MF, Totomoch-Serra A, Vargas-Alarcón G, Cruz-Robles D, Pellizzon OA, Cárdenas M. Síndrome de Andersen-Tawil: una revisión del diagnóstico genético y clínico con énfasis en sus manifestaciones cardíacas. Archivos de Cardiología de México. outubro de 2014;84(4):278–85.
18. Rebelo AR, Lopes A, Magalhães C, Sarmento J, Salgado M, Rebelo AR, et al. Síndroma de QT Longo: relato de caso de uma causa rara de síncope em idade pediátrica. Revista Portuguesa de Medicina Geral e Familiar. abril de 2021;37(2):170–3.
19. Sacilotto L, Olivetti NQS, Pisani CF, Wu TC, Hajjar LA, Melo SL de, et al. Peculiaridade dos Pacientes com Arritmias Hereditárias na Pandemia pela COVID-19. Arq Bras Cardiol. 6 de setembro de 2021;117:394–403.
20. Modoni A, Bianchi MLE, Vitulano N, Pagliarani S, Perna F, Sanna T, et al. Lack of Any Cardiac Involvement in a Patient with Andersen-Tawil Syndrome Associated with the c.574A→G Mutation in KCNJ2. Cardiology. 2011;120(4):200–3.
21. Jagodzińska M, Szperl M, Ponińska J, Kosiec A, Gajda R, Kukla P, et al. Coexistence of Andersen-Tawil Syndrome with Polymorphisms in hERG1 Gene (K897T) and SCN5A Gene (H558R) in One Family: ATS, K897t, H558r in One Family. Ann Noninvasive Electrocardiol. março de 2016;21(2):189–95.
22. Kukla P, Biernacka E, Baranchuk A, Jastrzebski M, Jagodzinska M. Electrocardiogram in Andersen-Tawil Syndrome. New Electrocardiographic Criteria for Diagnosis of Type-1 Andersen-Tawil Syndrome. CCR. 31 de maio de 2014;10(3):222–8.
23. Fonseca DJ, Vaz da Silva MJ. Canalopatias cardíacas: o papel das mutações nos canais de sódio. Rev Port Cardiol. 1o de fevereiro de 2018;37(2):179–99.24. Andrade C, Meireles J, Leão M, Silveira F. Clinodactyly and syndactyly – diagnostic clues for Andersen-Tawil syndrome. Arq Neuro-Psiquiatr. novembro de 2014;72(11):899–899.
Trabalho de conclusão de residência, apresentado ao curso de Clínica Médica do Hospital Adventista de Manaus, orientado pela Dra. Aline Brasil, como requisito parcial para obtenção do título de especialista em Clínica Médica.