REGISTRO DOI: 10.69849/revistaft/ra10202408311607
Giulliana Di Folco Machado
João Vitor dos Santos Coe
Carolina do Nascimento Cunha
João Pedro Rodrigues Monteiro da Silva
Orientadora: Carla Andrea Moreira Ferreira
ABSTRACT
Hereditary hemorrhagic telangiectasia, also known as Rendu-Osler-Weber syndrome, is an uncommon genetic disease characterized by arteriovenous malformations mainly in skin and mucous membranes, but also in visceral organs, lungs and even central nervous system, causing intermittent hemorrhagic episodes, some of them potentially severe. Their main consequence is iron deficiency anemia. Parvovirus b19 is a DNA virus with proerythroblast tropism whose infection is generally asymptomatic, but might cause cutaneous manifestations, such as infectious erythema, articular symptoms and medullary aplasia, rarely seen in immunocompetent individuals. The aim of this study is to demonstrate the association of these two diseases and their potential correlation. In this report we describe a 54 years old woman with a history of epistaxis since childhood, who presented worsening hemorrhagic episodes, melena, dyspnea at rest and pancytopenia. During investigation the diagnosis of Rendu-Osler-Weber syndrome was made, and posteriorly it was identified that her clinical worsening and pancytopenia was caused by medullary aplasia associated to parvovirus b19infection.
Keywords: Telangiectasia, Hereditary Hemorrhagic; Parvovirus B19, Human; Pancytopenia; Case reports.
RESUMO
A telangiectasia hereditária hemorrágica, ou síndrome de Rendu-Osler-Weber, é uma doença genética incomum que se caracteriza por malformações arteriovenosas principalmente em pele e mucosas, mas também acomete órgãos viscerais, pulmões e até o sistema nervoso central, cursando com episódios hemorrágicos constantes e potencialmente graves, tendo como consequência principal anemia por deficiência de ferro. O parvovírus b19 é um DNA vírus com tropismo por pró-eritroblastos cuja infecção é geralmente assintomática, porém, pode levar a manifestações cutâneas como eritema infeccioso, articulares e aplasia medular, sendo essa última rara em indivíduos imunocompetentes. O objetivo deste estudo é demonstrar a associação dessas duas doenças e sua potencial correlação. Descrevemos uma paciente do sexo feminino de 54 anos com quadro de epistaxes e anemia desde a infância, que se apresentou com piora do quadro hemorrágico, melena, dispneia em repouso e pancitopenia. Durante sua investigação foi feito o diagnóstico de síndrome de Rendu-Osler-Weber, e posteriormente identificou-se que a causa da piora clínica e pancitopenia foi a aplasia medular associada ao parvovírus b19.
Descritores: Telangiectasia Hemorrágica Hereditária; Parvovirus B19 Humano; Pancitopenia; Relatos de Casos.
INTRODUÇÃO
A telangiectasia hereditária hemorrágica (THH), também conhecida como síndrome de Rendu-Osler-Weber, é uma doença genética pouco comum, caracterizada por malformações arteriovenosas em mucosas, órgãos viscerais e no sistema nervoso central, ocasionando quadros de epistaxes, sangramento gastrointestinal, anemia ferropriva e telangiectasias (1-2).
O diagnóstico de THH é feito pelos Critérios de Curaçao: 1) epistaxe recorrente volumosa; 2) telangiectasias características; 3) malformações arteriovenosas ou telangiectasias viscerais e 4) família de primeiro grau portador da doença. Com 3 ou 4 dos critérios preenchidos, o paciente é classificado com diagnóstico definitivo de síndrome de Rendu-Osler-Weber (3-4).
O parvovírus B19, é transmitido por um Erythrovirus, da família Parvoviridae, a partir de gotículas de saliva (6). Este DNA vírus possui tropismo pelas células progenitoras dos eritrócitos, ocasionando uma diminuição ou até mesmo o desaparecimento dos reticulócitos mensuráveis, principalmente em indivíduos imunocomprometidos (7-8).
O paciente geralmente não apresenta sintomas, porém a doença pode cursar com artropatia, síndrome das luvas e meias, ocasionada por erupções papulares purpúricas e petequiais das mãos e pés, principalmente em crianças (9). Em indivíduos imunocomprometidos, existe uma maior chance de ocorrer a criseaplástica transitória.
Relatamos o caso de uma paciente que foi internada em nossa instituição para investigação de pancitopenia e episódios hemorrágicos. Sendo diagnosticada com THH e evidenciada aplasia medular devido infecção pelo parvovírus B19, uma associação não documentada em outros estudos anteriormente.
RELATO DE CASO
Paciente de 54 anos, sexo feminino, branca, brasileira, natural de Petrópolis (RJ), transferida de Unidade de Pronto Atendimento (UPA) ao Hospital Alcides Carneiro (HAC) para investigação diagnóstica de quadro de fraqueza, dispneia, melena e epistaxes.
Paciente relatou que desde a infância apresentava quadros de epistaxe volumosa recorrente e astenia, com prejuízo ao seu desenvolvimento psicomotor. Aos 15 anos de idade decide buscar ajuda médica e é diagnosticada com anemia ferropriva e iniciando reposição oral de ferro, porém sem elucidação etiológica ou redução dos episódios de epistaxe. Há 2 anos o quadro evoluiu para dispneia aos mínimos esforços associada a aumento dos episódios hemorrágicos, com presença de melena, e há um mês procurou pronto socorro, sendo internada devido a anemia severa, dispneia importante e redução da oximetria de pulso.
Em relação a sua história patológica pregressa, foi diagnosticada com hipertensão arterial sistêmica e asma brônquica aos 22 anos, diabetes melito tipo II aos 41 anos e doença arterial coronariana aos 52 anos, sem história de tabagismo ou etilismo. Aos 20 anos teve sua primeira gestação, porém sofreu aborto espontâneo devido a sangramento no 1º trimestre. Nas demais gestações não houveram intercorrências. Seus antecedentes familiares incluem 2 tios maternos que faleceram por cirrose hepática sem história de etilismo, filha e neta com epistaxes volumosas desde a infância.
Ao exame físico, apresentava-se hipocorada 3+/4+, havia presença de telangiectasias e petéquias em mucosa oral e nasofaríngea (figura 1), ausculta pulmonar com murmúrio vesicular universalmente reduzido, ausculta cardíaca com sopro sistólico pancardíaco, saturação pela oximetria de pulso de 88% em ar ambiente e taquicardia, sem mais alterações.
Foram solicitados exames laboratoriais na admissão, com os seguintes resultados: hemoglobina 6,3 g/dL; hematócrito 23,9 %; hemácias 3,47 x 1012/L; leucócitos 3,65 x 109/L; plaquetas 57 x 109/L; ureia 44,1 mg/dL; creatinina 1,36 mg/dL; ferro sérico 23,26 mcg/dL; ferritina 36,7 ng/mL, transferrina 255 mg/dL, sem mais alterações. Foram solicitadas tomografias com contraste e endoscopia digestiva alta (EDA) para pesquisa de malformações arteriovenosas e investigação de anemia ferropriva. A tomografia torácica mostrou aglomerado de vasos dilatados e serpenginosos em base pulmonar esquerda (figura 2), a abdominal hepatoesplenomegalia associada a circulação colateral periesplênica, com sinais de hipertensão portal, fígado com contorno irregular e hipertrofia do lobo quadrado, indicando hepatopatia crônica. EDA demonstrou múltiplas angiodisplasias gástricas e bulbo duodenais. Os resultados desses exames permitiram o diagnóstico na paciente de THH, com anemia ferropriva associada. Iniciada reposição de ferro venoso para tratamento e hemotransfusão com concentrado de hemácias.
Simultaneamente, prosseguiu-se a investigação da pancitopenia apresentada pela paciente. Foram solicitadas sorologias para possíveis vírus causadores de pancitopenia, além da realização de mielograma. No primeiro exame, houve positividade sorológica para Parvovírus B19 (IgM reagente 4,23 e IgG reagente 64,65). No mielograma, todos os setores encontravam-se bem apresentados, com aumento do setor eritróide e presença de pró-eritroblastos gigantes (figura 3). Assim, foi possível a elucidação diagnóstica adequada para a pancitopenia. Em síntese: trata-se de paciente com THH e anemia ferropriva associada, com descompensação clínica devido a aplasia medular causada pela infecção do parvovírus B19.
DISCUSSÃO
A THH é uma doença descrita no século XIX como uma combinação de epistaxes, sangramento gastrointestinal, anemia ferropriva e telangiectasias, tendo incidência de um caso em 5000-8000 indivíduos. Hoje, sabe-se que essas alterações advém de malformações arteriovenosas que também podem ocorrer em órgãos viscerais e até no sistema nervoso central (1).
A doença tem origem genética, de caráter autossômico dominante, causada por mutações nas proteínas angiogênicas TGF-β: ACVRL1, ENG e SMAD4. Suas manifestações clínicas mais comuns são telangiectasias e petéquias em mucosa oral e pele, entretanto, até 30% dos pacientes têm alterações vasculares em diversos outros órgãos: angiodisplasias gastrointestinais, malformações arteriovenosas pulmonares, cerebrais, espinhais e hepáticas (causando hipertensão portal e sobrecarga de volume no coração) (1-2).
A perda de sangue crônica sofrida por esses pacientes frequentemente leva a anemia ferropriva, e até 5% deles podem ter hemorragias severas das mucosas ou gastrointestinais. O diagnóstico de THH é feito pelos Critérios de Curaçao: 1) epistaxe recorrente volumosa; 2) telangiectasias características; 3) malformações arteriovenosas ou telangiectasias viscerais e 4) família de primeiro grau portador da doença. Com 3 ou 4 dos critérios preenchidos, o paciente é classificado como portador da síndrome de Rendu-Osler-Weber, exatamente o caso da paciente em estudo. Testes genéticos em familiares podem ser feitos em caso de incerteza diagnóstica, embora não haja recomendação forte nos principais estudos e guidelines (3-4).
O manejo da síndrome visa prevenir sangramentos e reduzir suas complicações. Para prevenção de epistaxe é recomendada umidificação nasal frequente, e seu tratamento pode requerer terapia ablativa e, em casos mais graves, anti-fibrinolíticos. Angiodisplasias gastrointestinais podem ser tratadas com terapia endoscópica local. A embolização das malformações arteriovenosas dos demais locais depende do tamanho, sintomas e riscos de complicações, devendo ser discutida com o especialista adequado. A anemia ferropriva deve ser tratada, mesmo em casos assintomáticos, com preferência para via oral. As anemias graves associadas à instabilidade hemodinâmica podem requerer uso de concentrados de hemácias. Por fim, novos tratamentos como anticorpos anti-VEGF ainda estão em estudo e podem ser considerados em pacientes refratários às terapias acima mencionadas (3-5).
Indivíduos imunocomprometidos ou com alterações na série vermelha, como aqueles acometidos pela THH, tem aumentada susceptibilidade a apresentar crises de aplasia medular devido ao parvovírus B19, algo raro em indivíduos saudáveis. Essa virose é transmitida por um Erythrovirus, da família Parvoviridae, a partir de gotículas de saliva e possui um período de incubação de 4 a 14 dias podendo chegar até 21 dias, sendo mais comumente encontrado em crianças (6). Este DNA vírus possui tropismo pelas células progenitoras dos eritrócitos, devido a associação do receptor globosídeo P e da, ocasionando uma diminuição ou até mesmo o desaparecimento dos reticulócitos mensuráveis (7-8).
O quadro da virose geralmente é assintomático, porém o paciente pode apresentar eritema infeccioso e artropatia. Os indivíduos imunocomprometidos, como portadores de hemoglobinopatias e anemias hemolíticas hereditárias, possuem maior chance de apresentar uma crise aplástica transitória. Esta complicação é definida por um um episódio transitório de aplasia pura das células vermelhas, com ausência virtual de precursores eritróides na medula óssea e dos reticulócitos na circulação, gerada pela hemólise crônica e/ou rápido turnover de eritrócitos. Os sintomas cursam com febre, mialgia, prostração, palidez, fadiga, e uma taquicardia secundária à anemia.
Esse DNA vírus, cujo o receptor, globosídeo (Gb4) ou antígeno P, é um glicoesfingolipídio neutro de membranas, está presente nas células da linhagem vermelha em grande quantidade, além de outros onze tecidos. A medula se torna uma grande reserva viral, pois as células progenitoras eritróides possuem o antígeno P e a integrina correceptora α5β1. Essa associação facilita a replicação viral no interior das células, que como consequência gera a interrupção da eritropoiese (9).
O diagnóstico da virose é feito pela sorologia positiva da parvovirose B19 e pode estar associado a reticulocitopenia, leucopenia, trombocitopenia e diminuição da hemoglobina. A crise aplásica é definida por uma hemoglobina menor que 2. Também é possível observar no exame da medula óssea aplasia grave da linhagem eritróide, surgindo por vezes, pró eritroblastos gigantes com inclusões virais (7). A doença é autolimitada, por isso o tratamento é sintomático e se necessário realização de transfusão sanguínea na crise aplásica (10).
CONCLUSÃO
Pacientes que convivem com a THH estão suscetíveis a apresentarem anemia ferropriva devido aos sangramentos constantes, e diversos fatores podem causar a descompensação clínico-hematológica. Nesse estudo relatamos a associação não antes relatada entre a THH e a aplasia medular causada pelo parvovírus B19, uma complicação que está mais bem descrita em pacientes imunocomprometidos. Por serem ambas doenças incomuns não há estudos que indiquem o grau de associação entre elas. Esse caso ilustra a importância do diagnóstico clínico da THH, que deve ser feito o mais precoce possível, e suas repercussões sistêmicas, além de demonstrar como doenças que afetam a hematopoiese podem causar complicações potencialmente graves. Há necessidade de novos estudos que elucidem a fisiopatologia dessas doenças e sua associação.
REFERÊNCIAS
1. Begbie ME, Wallace GM, Shovlin CL. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgrad Med J. 2003;79(927):18-24.
2. Burrows PE. Angioarchitecture of Hereditary Arteriovenous Malformations. Semin Intervent Radiol. 2017;34(3):250-257.
3. Kritharis A, Al-Samkari H, Kuter DJ. Hereditary hemorrhagic telangiectasia: diagnosis and management from the hematologist’s perspective. Haematologica. 2018;103(9):1433-1443.
4. Shovlin CL, Buscarini E, Sabbà C, et al. The European Rare Disease Network for HHT Frameworks for management of hereditary haemorrhagic telangiectasia in general and speciality care. Eur J Med Genet. 2022;65(1):104370.
5. Faughnan ME, Mager JJ, Hetts SW, et al. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Ann Intern Med. 2020;173(12):989-1001.
6. Cossart YE, Field AM, Cant B, Widdows D. Parvovirus-like particles in human sera. Lancet. 1975;1(7898):72-3.
7. Garcia SO, Pereira J, Sanabani S, Neto WK, Sabino EC. Doenças hematológicas associadas ao eritrovírus. Rev Bras Hemato Hemoter. 2009;31(4):285-90.
8. Weigel-Kelley KA, Yoder MC, Srivastava A. Alpha 5β1 integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of β1 integrin for viral entry. Blood. 2003; 102(12):3927-33.
9. Pinto MIM, Machado DM. Parvoviroses. In: Farhat CK, Carvalho ES, Carvalho LHF, Succi RCM, eds. Infectologia Pediátrica. 2a ed. Atheneu; 1998. p. 478-80.
10. Brown KE, et al. Parvovirus B19. In: Bennett JE, et al. Mandell, Douglas and Bennett’s. Principals and Practice of Infectious Diseases. 8th ed. Philadelphia, PA: Churchill Livingstone Elsevier. 2015. p. 1840-1847.
FIGURAS E TABELAS
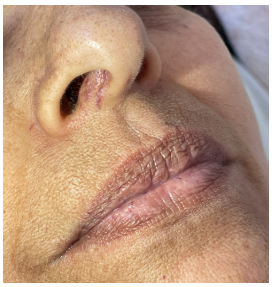
Figura 1. Telangiectasias em mucosa nasal. Algumas petéquias podem ser observadas no lábio inferior.
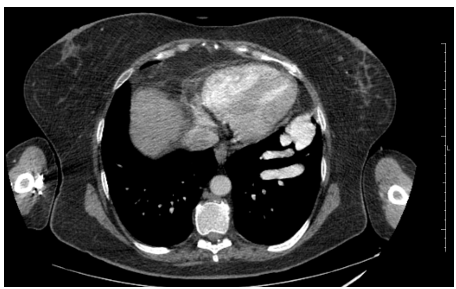
Figura 2. Aglomerado de vasos dilatados e serpenginosos na base pulmonar esquerda.
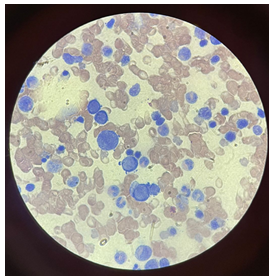
Figura 3. Mielograma demonstrando pró-eritroblastos gigantes com nucléolo evidente, compatível com infecção por parvovírus b19.
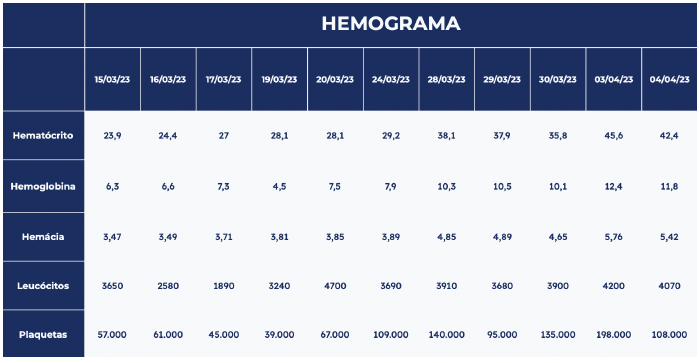
Tabela 1. Evolução dos parâmetros hematológicos da paciente. Nota-se uma piora progressiva da pancitopenia com posterior recuperação das séries simultaneamente a estabilização clínica.