DUNNIGAN SYNDROME OR FAMILIAL PARTIAL LIPODYSTROPHY TYPE 2 (FPDL2) REPORT OF TWO CASES
REGISTRO DOI: 10.69849/revistaft/cs10202408230052
Flávia Mazoti Saturi
Thalita Ferrari dos Santos
Frederico Alonso
Resumo
A Síndrome de Dunnigan, ou Lipodistrofia Parcial Familiar Tipo 2 (LPF2), é uma doença genética rara que provoca a perda seletiva e progressiva de tecido adiposo subcutâneo, geralmente após a puberdade. O tecido adiposo, um importante órgão endócrino, regula a homeostase energética, o metabolismo lipídico e hormonal, estando associado a várias condições metabólicas como obesidade, diabetes mellitus tipo 2 (DM2) lipodistrofias e doenças cardiovasculares. A lipodistrofia é classificada como congênita e adquirida de acordo com a sua origem, e generalizada e parcial, conforme seu acometimento corporal. (LEÃO et al., 2011).
A LPF2 é uma condição rara, com uma prevalência global estimada de 1 em cada 10 milhões de pessoas. A literatura aponta diferentes taxas de prevalência em diversos países. Nos Estados Unidos e na Noruega, a prevalência é de 1 em 10 milhões e 1 em 1 milhão de pessoas, respectivamente. No Brasil, a maior prevalência é observada no estado do Rio Grande do Norte, no Nordeste. Com uma proporção de 1 em 28 mil pessoas. Pesquisas genealógicas sobre as famílias que colonizaram essa região sugerem que a alta prevalência da lipodistrofia congênita está relacionada à origem portuguesa, além da grande quantidade de casamentos consanguíneos na região (BISPO & FREITAS, 2023).
Este trabalho, por meio do relato de dois casos clínicos, objetiva compreender os aspectos clínicos, genéticos e psicossociais da LPF2 destacando suas manifestações, diagnóstico e opções terapêuticas.
Palavras-chave: Lipodistrofia / lipodistrofia parcial familiar / genética humana / DEXA (Dual-Energy X-ray Absorptiometry)
1. INTRODUÇÃO
A Síndrome de Dunnigan, também conhecida como Lipodistrofia Parcial Tipo 2 (LPF2), é uma condição rara caracterizada pela perda seletiva e progressiva de tecido adiposo subcutâneo, geralmente iniciada após a puberdade. O tecido adiposo, é um órgão endócrino de extrema importância na homeostase energética, metabolismo lipídico e regulação hormonal, sendo esse diretamente ligado à diversas condições metabólicas, sendo elas, a obesidade, DM2, lipodistrofias e doenças cardiovasculares.
Existem dois tipos principais de tecido adiposo, o branco (TAB) e o marrom (TAM), o TAB é basicamente responsável pelo armazenamento de energia na forma de triglicerídeos, enquanto o TAM está envolvido na termogênese, um processo que gera calor a partir da queima de ácidos graxos (RUTKOWSKA et al., 2022). O tecido adiposo apresenta diversas funções endócrinas importantes. Os adipócitos secretam adipocinas, dentre elas adiponectina e leptina, que regulam o metabolismo da glicose, sensibilidade à insulina, inflamação e outros processos fisiológicos.
A LPF2 foi descrita pela primeira vez em 1974, pelo médico David Dunnigan apresenta um padrão de herança autossômico dominante, frequentemente resultante de mutações do tipo missense (sentido trocado) no gene LMNA, que codifica as proteínas da lâmina A e C. As lâminas são proteínas longas e filamentosas que compõem a lâmina-nuclear, as quais envolvem internamente o núcleo e interagem com a cromatina, regulando a replicação e o reparo do DNA, a transcrição e a organização da cromatina. O gene LMNA está localizado na região 22 do braço longo do cromossomo 1 e contém 12 éxons (SILVA, LIMA & CAMPOS, 2022).
Este trabalho tem como objetivo descrever os aspectos clínicos, genéticos e psicossociais da FPLD2 através do relato de dois casos. É fundamental compreender a função do tecido adiposo na patogênese desta condição para o reconhecimento de suas manifestações clínicas. Através de um diagnóstico, é possível intervir na prevenção de doenças cardiovasculares que podem se associar à LPF2.
2. FUNDAMENTAÇÃO TEÓRICA OU REVISÃO DA LITERATURA
2.1 FISIOLOGIA DO TECIDO ADIPOSO
O tecido adiposo é um componente vital do corpo humano, é composto por células especializadas no armazenamento de lipídios na forma de triacilglicerol, chamadas adipócitos, além de matriz extracelular e outros tipos de células, como fibroblastos, macrófagos e células endotelias. Consiste no principal reservatório energético do organismo, além disso, tem como atribuições o isolamento térmico, proteção de órgãos internos, função endócrina, regulação da modulação do metabolismo lipídico. Pode ser classificado em dois tipos principais, o tecido adiposo branco (TAB) e o tecido adiposo marrom (TAM) (FONSECA-ALANIZ et al., 2006). Os adipócitos do TAM contêm várias pequenas gotículas lipídicas e numerosas mitocôndrias ricas em UCP1 (proteína desaclopadora 1) utiliza a energia liberada pela oxidação de metabólitos para gerar calor, portanto é um tecido especializado na termogênese. É mais abundante em recém-nascidos e em menor quantidade em adultos, localizado principalmente em região supraclavicular, pescoço e ao longo da coluna vertebral.
O TAB é caracterizado por possuir uma grande gotícula lipídica e um núcleo periférico é encontrado predominantemente no tecido subcutâneo e ao redor dos órgãos internos, é o principal local de armazenamento de energia na forma de triglicerídeos, que podem ser convertidos em ácidos graxos e glicerol, e utilizados como fonte de energia. Tem papel importante como isolante térmico e protetor mecânico contra traumatismos externos. Ademais, o TAB é um órgão envolvido em processos endócrinos, capaz de secretar adipocinas, que incluem a leptina, adiponectina, resistina, fator de necrose tumoral alfa (TNF-a), dentre outros. A leptina regula o balanço energético, atuando sobre neurônios hipotalâmicos com consequente inibição da ingestão alimentar e aumento do gasto energético total, ainda inibe a expressão do neuropeptídeo Y, envolvido no mecanismo de aumento da ingestão alimentar. A adiponectina aumenta a sensibilidade à insulina e possui efeitos anti-inflamatórios e anti-ateroscleróticos, com correlação inversa entre níveis circulantes do hormônio e risco de obesidade, resistência à insulina e outras doenças cardiovasculares (FONSECA-ALANIZ et al., 2007).
O TAB divide-se em dois tipos: o tecido adiposo visceral e o tecido adiposo subcutâneo. O primeiro está localizado na cavidade abdominal, em torno dos órgãos internos como o fígado, o pâncreas e os intestinos. Devido à sua proximidade com os órgãos internos, a gordura visceral tem um impacto direto no metabolismo, liberando ácidos gordos diretamente no fígado, o que pode contribuir para problemas metabólicos. Por outro lado, o tecido adiposo subcutâneo encontra-se logo abaixo da pele e está distribuído por todo o corpo, sendo mais concentrado em áreas como o abdômen, quadris, coxas e nádegas. A função principal deste tipo de gordura é servir como isolante térmico e reserva de energia. Embora o tecido adiposo subcutâneo seja mais visível e esteja associado à estética corporal, ele é menos prejudicial para a saúde em comparação com a gordura visceral (FONSECA-ALANIZ et al., 2006).
2.2 FISIOPATOLOGIA E CLASSIFICAÇÃO
As lipodistrofias são doenças genéticas caracterizadas pela de tecido adiposo subcutâneo de forma generalizada ou localizada, sem que haja desnutrição ou estado catabólico. É um grupo heterogêneo de doenças, com ampla apresentação, podendo apresentar resistência à insulina, DM2, hipertrigliceridemia, hipertrofia muscular, acantose nigricans e características faciais peculiares. Podem ser classificadas em congênita ou adquirida. As formas adquiridas, não são associadas a mutações genéticas, e a sua fisiopatologia envolve mecanismos autoimunes, (Referência Lipodystrophy for the diabetologist – 2022 – Natália mandou) As lipodistrofias congênitas são determinadas por mutações específicas. A Lipodistrofia Generalizada Congênita (LGC) tem como protótipo a Síndrome de Berardinelli-Seip, condição autossômica recessiva, com mutação em genes como AGPAT2 e BSCL2, geralmente diagnosticada logo após o nascimento, caracterizada pela ausência ou diminuição acentuada do tecido adiposo corporal, acompanhada de resistência insulínica, hipertrigliceridemia e acantose nigricans (LEÃO et al., 2011).
A LPF2 é causada por mutações no gene LMNA, geralmente pela substituição de um único aminoácido na proteína (SILVA, LIMA & CAMPOS, 2022). Algumas das mutações específicas mais comuns no LMNA associadas à LPF2 incluem: R482W (arginina para triptofano na posição 482), que é a mais frequente; R482Q (arginina para glutamina na posição 482), com variações ligeiramente diferentes no fenótipo; e R482L (arginina para leucina na posição 482) (MONTEIRO et al., 2012).
2.3 MANIFESTAÇÕES CLÍNICAS
A LPF2 se caracteriza por perda progressiva de tecido adiposo subcutâneo, membros inferiores e superiores, glúteos, abdômen e tronco, com consequente acúmulo de gordura em regiões como pescoço, face e região intra-abdominal (imagem 19). Essa composição corporal diversa confere aparência de hipertrofia muscular, flebomegalia nas extremidades e acúmulo de gordura na face e região cervical. Essas alterações na aparência física podem impactar negativamente a vida social dos indivíduos, e levar a problemas de autoimagem e autoestima, além de afetar a saúde mental dos pacientes, podendo resultar em depressão e ansiedade.
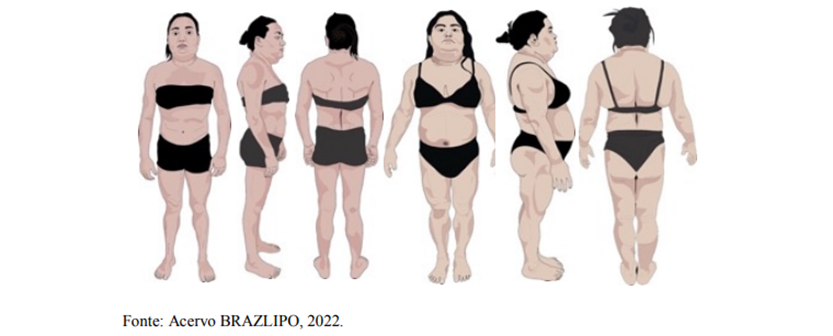
Figura 1 (Fenótipos de LPF2 com mutações do gene LMNA)
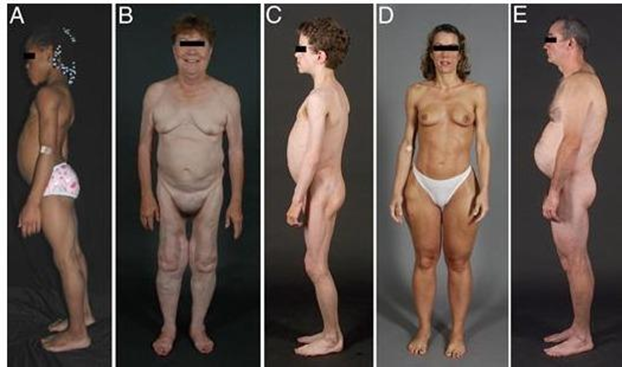
Figura 2 – Clínicas de pacientes com vários tipos de lipodistrofias (A: lipodistrofia congênita generalizada – Berardinelli-Seip / B: LPF2 – Dunnigan / C: lipodistrofia generalizada adquirida / D: lipodistrofia parcial adquirida – Barraquer-Simons / E: paciente com HIV com lipodistrofia induzida por terapia antirretroviral
Os pacientes podem apresentar exames laboratoriais típicos da doença, como hiperinsulinemia, resistência à insulina, glicemia e hemoglobina glicada elevada, dismorfismo lipídico, hiperandrogenismo, marcadores de inflamação e alterações hepáticas.
Ao comparar mulheres com LPF2 e mulheres da população em geral, as mulheres com LPF2 são mais gravemente afetadas com complicações metabólicas e doenças cardiovasculares.
2.4 DIAGNÓSTICO
O diagnóstico dessa doença pode ser desafiador devido à sua apresentação clínica variável, embora característica, e à sobreposição de sintomas com outras condições metabólicas como resistência à insulina e síndrome metabólica. Baseia-se em uma avaliação sequencial de características clínicas, dados antropométricos, exames de composição corporal e testes genéticos.
Observa-se os aspectos fenotípicos, como a perda progressiva de gordura subcutânea nas extremidades e tronco, combinada com acúmulo de gordura em região cervical e genital. Esse padrão é acompanhado por complicações metabólicas, como resistência à insulina, diabetes tipo 2 e dislipidemia. Além disso, os dados antropométricos são avaliados, com ênfase nas medidas de pregas subcutâneas, IMC e o índice de Köb.
A confirmação pode ser suportada por outros métodos como a DEXA (do inglês Dual-Energy X-ray Absorptiometry), técnica de imagem não invasiva, que tem sido utilizada para estimativa da composição corporal. No contexto da FPLD2, seu papel se destaca pela capacidade de fornecer informações detalhadas sobre a distribuição e medição quantitativa de gordura corporal, auxiliando no diagnóstico e monitoramento da doença. Neste estudo, as medições DEXA da distribuição de gordura ajudaram a melhor caracterizar o padrão específico de lipodistrofia presente.
Finalmente, o estudo genético confirma o diagnóstico com a identificação de mutações no gene LMNA (como R482W, R482Q ou R482L). A presença de uma dessas mutações confirma a LPF2 e auxilia no aconselhamento genético.
2.5 DIAGNÓSTICOS DIFERENCIAIS
Ao considerar o diagnóstico de LPF2, é essencial também considerar outras condições que podem apresentar características semelhantes. É importante distinguir a LPF2 de outras lipodistrofias), que envolve perda difusa de tecido adiposo em todo o corpo desde o nascimento ou primeira infância; a Lipodistrofia Parcial Adquirida (Síndrome de Barraquer-Simons), que se inicia na infância ou puberdade, com perda de gordura que frequentemente se inicia em face e progride para o tronco, com acúmulo em coxas e quadris, e é muitas vezes associada a doenças autoimunes; a Lipodistrofia Generalizada Adquirida (Síndrome de Lawrence) determinada pela perda de tecido adiposo em todo o corpo, frequentemente associada a doenças autoimunes ou em contexto de HIV/AIDS; a Lipodistrofia Parcial Familiar do Tipo 1 (Síndrome de Köbberling), caracterizada por perda de gordura em membros inferiores, enquanto a parte corporal superior retém gordura. (VERAS, 2023).
Outro diagnóstico diferencial que deve ser levado em conta é a Síndrome de Cushing, caracterizada pelo excesso de cortisol, marcada pela obesidade central, resistência à insulina, hiperandrogenismo, “face em lua cheia”, fraqueza muscular, pele fina e estrias violáceas.
A Síndrome dos Ovários Policísticos (SOP) é um importante diagnóstico diferencial devido suas características de hiperandrogenismo com índices de Ferriman-Gallwey variáveis, irregularidades menstruais, resistência à insulina, ovários policísticos e obesidade.
A Síndrome Metabólica é um conjunto de condições que ocorrem aumentando o risco cardiovascular dos pacientes, ela pode ser tanto um diagnóstico diferencial quanto parte da clínica da LPF2, devido à sobreposição de características metabólicas apresentadas nessa doença. Seus critérios diagnósticos, segundo o International Diabetes Federation (IDF), consistem na medida da circunferência abdominal, pressão arterial, triglicerídeos, colesterol HDL e glicemia de jejum (SANTOS, SCHRANK & KUPFER, 2009)
2.6 TRATAMENTO
O tratamento eficaz requer uma abordagem multidisciplinar, envolvendo endocrinologistas, cardiologistas, nutricionistas e psicólogos. A terapêutica atual direciona-se ao controle das alterações metabólicas, prevenção e tratamento de complicações relacionadas à LPF2, como diabetes mellitus, dislipidemia, hepatopatia, pancreatite, HAS, doenças cardiovasculares e SOP.
Modificações no estilo de vida, como dieta saudável rica em fibras, com baixo teor glicêmico e gorduras saturadas é crucial para o controle do metabolismo, além de estímulo à prática de exercícios físicos para diminuir a resistência à insulina e controle do peso. Porém, ainda não há estudos que avaliem o impacto dessas medidas em pacientes com LPF2 (VERAS, 2023).
É essencial que haja monitoramento regular geral do paciente, como glicemia, perfil lipídico e função hepática, além de avaliação cardiovascular.
Frequentemente são prescritas terapias para tratar as alterações metabólicas, como metformina, tiazolidinedionas, inibidores de SGLT2, estatinas e fibratos, todavia elas têm eficácia limitada devido à natureza genética e complexa da doença. Esses medicamentos tratam os sintomas metabólicos, mas não corrigem a redistribuição anômala de gordura e as complicações metabólicas associadas à disfunção genética da LPF2. Portanto, seu impacto é restrito, e o manejo da LPF2 requer uma abordagem mais abrangente e personalizada. (VERAS, 2023).
A metformina é usada com terapia de primeira linha para melhorar a sensibilidade à insulina e reduzir os níveis glicêmicos. As tiazolidinedionas melhoram a captação de glicose pelo músculo e pelo tecido adiposo, contribuindo no aumento da sensibilidade à insulina. Os inibidores de SGLT2 ajudam no controle glicêmico e na redução de peso, oferecendo benefícios adicionais. Devido à elevada resistência insulínica, muitos desses pacientes podem ainda utilizar altas doses de insulina no manejo de diabetes. Estatinas e fibratos são comumente prescritos para controle de aumento de LDL e de triglicerídeos, respectivamente, constantemente encontrados nas lipodistrofias.
Estudos atuais com o uso de metreleptina, um análogo sintético do hormônio leptina, têm mostrado eficácia para melhora do fenótipo lipodistrófico e do controle metabólico em pacientes portadores de LPF2, contudo mais pesquisas são necessárias para elucidação de suas indicações (LAMBADIARI et al., 2021).
A cirurgia plástica pode também auxiliar na gestão dos aspectos físicos e estéticos associados à distribuição anômala de gordura que caracteriza essa condição genética. Embora o foco do manejo da LPF2 seja a abordagem das complicações metabólicas e cardiovasculares, muitos pacientes sofrem com problemas de ordem psicossocial relacionados à imagem corporal, tendo a cirurgia plástica papel de melhorar a aparência e autoestima dos pacientes.
Por fim, a educação dos pacientes e suas famílias a respeito da importância dessa condição e seu manejo proativo é crucial. Apoio psicológico e grupos de suporte podem ser benéficos para o enfrentamento das mudanças estéticas e metabólicas geradas por essa doença, podendo contribuir para a aceitação para lidar com os desafios emocionais associados à LPF2.
3. METODOLOGIA
O estudo foi realizado com duas pacientes diagnosticadas com LPF2. A coleta de dados clínicos foi realizada através de uma minuciosa revisão dos prontuários das pacientes. Para mais, o estudo foi facilitado por meio de discussões e revisões organizacionais pelo orientador Dr. Frederico Alonso Sabino, coordenador do curso de Medicina do Centro Universitário Municipal de Franca (Uni-FACEF).
Ademais, entrevistas individuais foram conduzidas com cada paciente para complementar as informações obtidas nos prontuários. Durante a anamnese, foram abordados, principalmente, histórico familiar de doenças metabólicas e repercussões biopsicossociais da condição.
As participantes do estudo tiveram a oportunidade de realizar o estudo genético e de composição corporal (DXA), tais dados foram cedidos pela Dra. Natália Rossin Guidorizzi, endocrinologista responsável pelo protocolo de avaliação de lipodistrofias.
O exame genético que permitiu o estudo de sequenciamento do gene LMNA, foi realizado pelo laboratório Mendelics. O método utilizado começa com a construção de bibliotecas de DNA usando a tecnologia Illumina e a captura de regiões alvo. O sequenciamento é realizado na plataforma Illumina NovaSeq 6000. As leituras de DNA são alinhadas à versão GRCh38 do genoma humano utilizando o software específico. As variantes e pequenas deleções (indels) são identificadas com o GATK versão 4, enquanto as variações do número de cópias (CNVs) são detectadas pelo ExomeDepth. A análise médica é personalizada com base nas informações clínicas do paciente. As frequências alélicas das variantes são comparadas com o banco de dados GnomAD. A classificação das variantes segue as diretrizes do American College of Medical Genetics and Genomics (ACMG). Este método tem algumas limitações, como a sensibilidade reduzida para detectar CNVs que envolvem apenas um ou dois éxons e a dificuldade em analisar genes com pseudogenes ou regiões de alta homologia. Além disso, não identifica anomalias cromossômicas complexas, como aneuploidias.
Este estudo foi conduzido em conformidade com as diretrizes éticas para pesquisa com seres humanos, assegurando a confidencialidade e o anonimato das pacientes. As participantes foram devidamente informadas sobre os objetivos, procedimentos e possíveis implicações dos testes. A pesquisa foi aprovada pelo Comitê de Ética em Pesquisa do Uni-FACEF, conforme número de protocolo 6.926.064.
4. ANÁLISE DE DADOS
CASO 1
Paciente F.G.F., feminina, 28 anos, foi encaminhada para avaliação endocrinológica, apresentando perda progressiva de gordura subcutânea em membros e tronco desde os 15 anos. Apresentou menarca aos 13 anos e amenorreia após. Aos 14 anos teve diagnóstico de SOP, sendo iniciado o uso de contraceptivo oral combinado (COC). Mãe apresenta Dislipidemia (DLP), Hipertensão Arterial Sistêmica (HAS) e redução de tecido adiposo nos membros superiores e inferiores, característica que também é evidente em vários membros da família, acentuada em duas primas maternas que já estão em investigação endocrinológica.
Exame físico:
PARÂMETROS VALORES PESO (kg) 52,100 ESTATURA (cm) 168 ÍNDICE DE MASSA CORPORAL (IMC) (kg/m²) 18,459 CIRCUNFERÊNCIA ABDOMINAL (cm) 71 PREGA SUBCUTÂNEA COXA (mm) 7 PREGA SUBCUTÂNEA TRÍCEPS (mm) 3,5 PREGA SUBCUTÂNEA SUBESCAPULAR (mm) 12 PREGA SUBCUTÂNEA PANTURRILHA (mm) 4 ÍNDICE DE KÖB 3
Tabela 1. Dados antropométricos e Índice de Köb
PA 130×80 mmHg e FC: 140 bpm. TAS escasso em membros superiores e membros inferiores, aparente hipertrofia muscular e flebomegalia, evidenciado por meio do índice de Köb, calculado através da razão entre dobra subcutânea subescapular e de panturrilha, que quando maior que 3,477 é altamente sugestivo da LPF1 ou Sindrome de Kobberling (GUILLÍN-AMARELLE et al., 2016).
Presença de acantose nigricans nas regiões cervical e axilar, pele seca e áspera. Cabelos com rarefação nucal e hirsutismo ausente (paciente depilada). Genitália feminina, sem clitorimegalia, grandes lábios volumosos em decorrência de deposição de TAS.
No momento, paciente estava em uso regular de Espironolactona 50 mg, Metformina XR 1000mg, Succinato de Metoprolol 50mg, Drosperinona 3mg + Etinilestradiol 0,02 mg.
Exames laboratoriais: glicose 82 mg/dL; insulina 45 mUI/ml (Ref: 2,6-24); hemoglobina glicada 4,7%; colesterol total 202 mg/dL; LDL 134 mg/dL; HDL 43 mg/dL; triglicérides 125 mg/dL TGO, TGP, GGT sem alterações; ferritina 504 ng/ml (Ref: 13-150) creatinina 0,8 mg/dL ; LH 17,27 mUI/ml; FSH 5,99 mUI/ml; prolactina 24,14 ng/mL; estradiol 35 pg/mL; SDHEA 252 pg/mL (Ref: 98-340); androstenediona 5,3 ng/ml (Ref: 0,3-3,3); testosterona total 146 ng/dL (Ref: 8-49); 17 OHP 1,55 ng/ml (Ref:10-120 ng/dL), TSH 3,04 mUI/ml; T4L 0,77 ng/dL; anti-TPO negativo (exames colhidos sem uso de COC) ECG taquicardia sinusal, 100 bpm, cocardiograma sem alterações; US abdome total presença de colecistolitíase; US transvaginal: útero normal, ovário direito com microcistos na periferia e parênquima hiper ecogênico, medindo 3,5×2,7×3,9cm e volume de 19cm². Ovário esquerdo com microcistos na periferia e parênquima hiper ecogênico, medindo 3,5×2,8×3,9cm e volume de 20cm². (Figura 3)
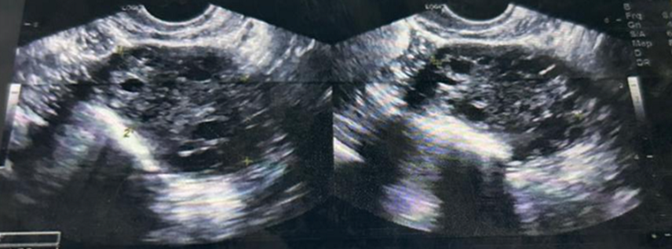
Figura 3 (US transvaginal – cistos ovarianos)
Com base nos achados clínicos, antropométricos e laboratoriais, foi suspeitado LPF2 e a paciente foi submetida ao estudo corporal com DEXA, que possibilitou análise de índices importantes para quantificação de lipodistrofia como o Índice Fat Mass Ratio (FMR), razão do percentual de gordura em tronco pelo percentual de gordura em perna, que recentemente foi sugerido um ponto de corte maior que 1,2 para indicar a LPF2 (VALERIO et al., 2012). O Índice PAR, razão entre massa gorda das pernas (g) por massa gorda total (g), também sugere LPF2 quando resultado <25% (BONNET et al., 2005). Além desses, o percentual isolado de gordura nas pernas <25% reforça escassez de tecido adiposo subcutâneo, característico de lipodistrofia.
PARÂMETROS VALORES GORDURA EM TROCO (%) 20 GORDURA EM PERNAS (%) 11,4 MASSA GORDA DAS PERNAS (g) 1.850 MASSA GORDA TOTAL (g) 9.387 PERCENTUAL ISOLADO DE GORDURA NAS PERNAS (%) 11,8 ÍNDICE FAT MASS RATIO – FMR 1,75 RAZÃO ENTRE MASSA GORA DAS PERNAS POR MASSA GORDA TOTAL – PAR 0,197
Tabela 2. Dados da DEXA e cálculos confirmatórios de escassez de tecido adiposo.
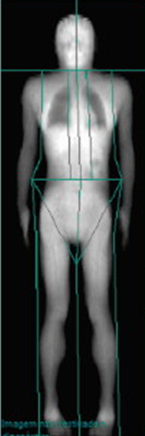
Figura 4 (DEXA)
No teste genético foi identificada, em heterozigose, no gene LMNA, variante promoveu a substituição do aminoácido arginina no códon 482 por triptofano (R482W), confirmando o diagnóstico de LPF2.
CASO 2
Paciente A.C.G.F., feminina, 54 anos, iniciou acompanhamento endocrinológico devido diagnóstico de LPF2 de sua filha e fenótipo semelhante ao dela. Relatou perda de gordura subcutânea durante a puberdade. Histórico de HAS há 25 anos, recentemente passou a apresentar glicemias elevadas.
Durante a investigação, paciente estava em uso regular de Espironolactona 25mg, Losartana 50mg, Metformina 1000mg, Atenolol 25mg.
Exame físico:
PARÂMETROS VALORES PESO (kg) 46,000 ESTATURA (cm) 147 ÍNDICE DE MASSA CORPORAL (IMC) (kg/m²) 21,143 CIRCUNFERÊNCIA ABDOMINAL (cm) 69 PREGA SUBCUTÂNEA COXA (mm) 5 PREGA SUBCUTÂNEA TRÍCEPS (mm) 3,5 PREGA SUBCUTÂNEA SUBESCAPULAR (mm) 21 PREGA SUBCUTÂNEA PANTURRILHA (mm) 3,5 ÍNDICE DE KÖB 6
Tabela 3. Dados antropométricos e Índice de Köb
PA:140x80mmHg e FC 80bpm. Perda de gordura subcutânea nos membros superiores e inferiores, hipertrofia muscular e presença importante de deposição de gordura mento (“duplo queixo”) e região pubiana. Acantose nigricans nas regiões cervical, axilar e inguinal. Exames laboratoriais: glicose 140mg/dL; insulina 66,4mUI/ml (Ref: 2,6-24); hemoglobina glicada 6,1% (Ref: <5,7%); colesterol total 177mg/dL; LDL 93mg/dL; HDL 40mg/dL; triglicérides 219mg/dL; TGO 98u/L(Ref: 10-35); TGP 88u/L (Ref: 7-45); GGT 68u/L (Ref: 5-36); FA 127u/L; ferritina 1,6ng/mL (Ref:10-290); creatinina 0,6 mg/dL; ureia 21 mg/dL; FSH 13,86iu/mL; TSH 3,7iu/mL; androstenediona 1,8ng/ml; testosterona total 12ng/dL; HBSAG não reagente; anti HBS, anti HCV e anti-HIV negativos; US de abdome sem alterações; US transvaginal apresentando útero de dimensões aumentadas com discreta alteração textural difusa do miométrio. Formação ecogênica em cavidade uterina podendo corresponder a pólipo. Cistos de retenção em colo uterino; US de abdome total, sugerindo nefrolitíase à direita, ECG com ritmo sinusal e eixo normoposicionado.
PARÂMETROS VALORES GORDURA EM TROCO (%) 31,2 GORDURA EM PERNAS (%) 14,5 MASSA GORDA DAS PERNAS (g) 1.753 MASSA GORDA TOTAL (g) 10.868 PERCENTUAL ISOLADO DE GORDURA NAS PERNAS (%) 15,1 ÍNDICE FAT MASS RATIO – FMR 2,15 RAZÃO ENTRE MASSA GORDA DAS PERNAS POR MASSA GORDA TOTAL – PAR 0,16
Tabela 4. Dados da DEXA e cálculos confirmatórios de escassez de tecido adiposo.
Ao analisar os parâmetros citados anteriormente para análise de confirmação de escassez de tecido adiposo subcutâneo, a paciente se enquadrou nos valores de referência diagnósticos. Apresentando, FMR maior que 1,2, índice de PAR < 25% e percentual isolado de gordura nas pernas menor que 25%, critérios estabelecidos em estudos prévios, reforçando a escassez de tecido adiposo subcutâneo característica.
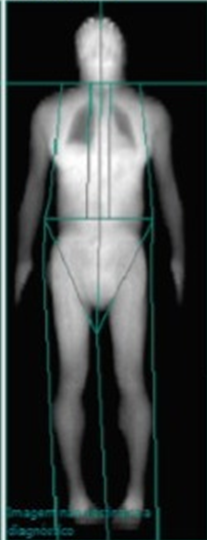
Figura 5 (DEXA)
No exame genético foi identificada em heterozigose, no gene LMNA variante que promoveu a substituição do aminoácido arginina no códon 482 por triptofano (R482W). Com essa análise, foi possível evidências que a mutação da paciente 1 é a mesma da paciente 2, possibilitando a confirmação do mesmo diagnóstico nas duas pacientes em discussão.
5. CONCLUSÃO/CONSIDERAÇÕES FINAIS
Em conclusão, este estudo ressalta a importância do diagnóstico precoce da LPF2 para a prevenção e tratamento dos distúrbios metabólicos potencialmente graves associados à condição. O fenótipo característico das pacientes afetadas, incluindo a perda do TAS, acantose nigricans, hipertrofia muscular aparente e acúmulo de gordura nos grandes lábios, alerta clínicos, endocrinologistas, cardiologistas e ginecologistas quanto ao reconhecimento da LPF2, dentre os pacientes que procuram atendimento por condições frequentes na população como SM e DM2, HAS, DLP, doença hepática gordurosa associada a disfunção metabólica e SOP (CALDAS et al., 2013).
É notável que ambos os casos ilustram a complexidade da LPF2, evidenciando diferença nos polos de complicação de acordo com a idade das pacientes, sendo que a paciente mais nova (28 anos) apresenta repercussões mais centradas na SOP e hiperandrogenismo, enquanto a paciente de 54 anos enfrenta desafios com resistência à insulina e hipertensão, refletindo um curso mais avançado da doença.
Ademais, desde a identificação da primeira mutação heterozigótica missense p.R482Q no éxon 8 do gene LMNA, a lista de mutações associadas à LPF2 tem se expandido rapidamente, com a maioria delas localizadas no códon 482. Os novos casos diagnosticados frequentemente apresentam essas mutações, com um fenótipo clássico de lipodistrofia e mínimo comprometimento de outros tecidos (CALDAS et al., 2013).
A compreensão detalhada das alterações clínicas, bioquímicas e genéticas da LPF2 pode contribuir significativamente para o entendimento dos mecanismos subjacentes à deterioração metabólica e resistência à insulina. Esse conhecimento é crucial não apenas para desenvolver novas terapias para condições raras como as lipodistrofias, mas também para abordar formas epidêmicas de resistência insulínica, como as observadas em pacientes com Síndrome Metabólica, cujos mecanismos genéticos ainda não estão completamente elucidados (LEÃO et al., 2011).
REFERÊNCIAS
1) Leão LM, Alencar RC, Rodrigues Gda C, Bouzas I, Gallo P, Rossini A. Lipodistrofia parcial familiar do tipo Dunnigan: atenção ao diagnóstico precoce [Dunnigan-type familial partial lipodystrophy: attention to precocious diagnosis]. Rev Bras Ginecol Obstet. 2011 Feb;33(2):99-103. Portuguese.
2) Bispo, Mônica & Freitas, Ana. (2023). Lipodistrofia generalizada congênita: uma revisão bibliográfica. Research, Society and Development. 12.
3) Rutkowska L, Salachna D, Lewandowski K, Lewiński A, Gach A. Familial Partial Lipodystrophy-Literature Review and Report of a Novel Variant in PPARG Expanding the Spectrum of Disease-Causing Alterations in FPLD3. Diagnostics (Basel). 2022 Apr 30;12(5):1122.
4) Fonseca-Alaniz MH, Takada J, Alonso-Vale MIC, Lima FB. O tecido adiposo como centro regulador do metabolismo. Arq Bras Endocrinol Metab [Internet]. 2006Apr;50(2):216–29.
5) Fonseca-Alaniz, M. H., Takada, J., Alonso-Vale, M. I. C., & Lima, F. B.. (2007). O tecido adiposo como órgão endócrino: da teoria à prática. Jornal De Pediatria, 83(5), S192–S203.
6) SILVA, M. A. da; LIMA, J. G.; CAMPOS, J. T. A. de M. Síndrome de Dunnigan: uma rara doença relacionada ao gene LMNA. Genética na Escola, São Paulo, v. 17, n. 2, p. 246–250, 2022.
7) Monteiro LZ, Foss-Freitas MC, Júnior Montenegro RM, Foss MC. Body fat distribution in women with familial partial lipodystrophy caused by mutation in the lamin A/C gene. Indian J Endocrinol Metab. 2012 Jan;16(1):136-8
8) VERAS, Victor Resende. Variabilidade clínica e genética na lipodistrofia parcial familiar. 2023. 96 f. Dissertação (Mestrado em Ciências Médicas) – Faculdade de Medicina, Universidade Federal do Ceará, Fortaleza, 2023.
9) Lambadiari V, Kountouri A, Maratou E, Liatis S, Dimitriadis GD, Karpe F. Case Report: Metreleptin Treatment in a Patient With a Novel Mutation for Familial Partial Lipodystrophy Type 3, Presenting With Uncontrolled Diabetes and Insulin Resistance. Front Endocrinol (Lausanne). 2021 Jun 8;12:684182.
10) Caldas D, Silva Júnior WS da, Simonetti JP, Costa EV da, Farias MLF de. [ARTIGO PARCIALMENTE RETRATADO] Avaliação bioquímica, hormonal e genética das famílias de duas pacientes brasileiras portadoras de lipodistrofia parcial familiar tipo 2. Arq Bras Endocrinol Metab [Internet]. 2013Nov;57(8):583–93
11) Guillín-Amarelle C, Sánchez-Iglesias S, Castro-Pais A, Rodriguez-Cañete L, Ordóñez-Mayán L, Pazos M, González-Méndez B, Rodríguez-García S, Casanueva FF, Fernández-Marmiesse A, Araújo-Vilar D. Type 1 familial partial lipodystrophy: understanding the Köbberling syndrome. Endocrine. 2016 Nov;54(2):411-421.
12) Valerio CM, Zajdenverg L, de Oliveira JE, Mory PB, Moyses RS, Godoy-Matos AF. Body composition study by dual-energy x-ray absorptiometry in familial partial lipodystrophy: finding new tools for an objective evaluation. Diabetol Metab Syndr. 2012 Aug 31;4(1):40. doi: 10.1186/1758-5996-4-40. Erratum in: Diabetol Metab Syndr. 2015 Mar 14;7:19.
13) Bonnet E, Delpierre C, Sommet A, Marion-Latard F, Hervé R, Aquilina C, Labau E, Obadia M, Marchou B, Massip P, Perret B, Bernard J. Total body composition by DXA of 241 HIV-negative men and 162 HIV-infected men: proposal of reference values for defining lipodystrophy. J Clin Densitom. 2005 Fall;8(3):287-92.
14) Garg A. Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96(11):3313-25.
15) Garg A. Vinaitheerthan M, Weatherall PT, Bowcock AM. Phenotypic heterogeneity in patients with familial partial lipodystrophy (Dunnigan variety) related to the site of missense mutations in lamin A/C gene. J Clin Endocrinol Metab. 2001;86:50-65.
16) Guidorizzi NR, Valerio CM, Viola LF, Veras VR, Fernandes VO, Lima GEDCP, Flor AC, Araújo JS, Gonçalves Muniz RB, Moreira RO, De Paula FJA, Zajdenverg L, Dantas JR, Godoy-Matos AF, Montenegro Júnior RM, Foss-Freitas MC. Comprehensive analysis of morbidity and mortality patterns in familial partial lipodystrophy patients: insights from a population study. Front Endocrinol (Lausanne). 2024 Jun 3;15:1359211.
17) Santos, Carlos, et al. “Artigo Original Análise Crítica Dos Critérios Da OMS, IDF E NCEP Para Síndrome Metabólica Em Pacientes Portadores de Diabetes Melito Tipo 1 Critical Analysis of WHO, IDF and NCEP Criteria for Metabolic Syndrome among Patients with Type 1 Diabetes Mellitus.” Arq Bras Endocrinol Metab, vol. 53, no. 9, 2009, pp. 1096–102