REGISTRO DOI: 10.5281/zenodo.10542447
Carolina Marques Ribeiro Pessoa*
Deborah Regina Cavalcante da Silva*
Ianny Karol Costa e Silva*
Lais Nadille Lins Coelho*
Camila Arcanjo Alves de Lima**
RESUMO
Introdução: A Síndrome de Beckwith-Wiedemann (SBW) é uma síndrome multissistêmica congênita descrita no início da década de 1960 pelo patologista pediátrico americano John Bruce Beckwith e pelo pediatra alemão Hans-Rudolf Wiedemann. É caracterizada por hipoglicemia, macroglossia, onfalocele, gigantismo, visceromegalia, idade óssea avançada, displasia renal, entre outros. Vários mecanismos genéticos diferentes podem causar a síndrome e o diagnóstico é realizado geralmente no período pós-natal. Objetivo: Relatar o caso clínico de um paciente com SBW. Relato de caso: Paciente do sexo masculino, 2 meses, com o diagnóstico clínico de SBW, que apresentou intercorrências como onfalocele e hérnia de parede umbilical, macroglossia, hipoglicemia, dispneia, cianose, apneia obstrutiva, pneumonia por bronco aspiração, genitália com macrofalus, criptorquidia bilateral, icterícia e cardiomegalia com coração em bota, desde o nascimento decorrentes da mesma. Discussão: A SBW é conhecida por super crescimento. Os pacientes podem apresentar diversas condições clínicas que necessitarão de abordagem multiprofissional, dentre elas, a indicação de redução da macroglossia, observada em 50% dos casos. O objetivo é prevenir complicações e melhorar a qualidade de vida. O prognóstico é favorável, com alta sobrevida no primeiro ano. Conclusão: O diagnóstico de pacientes com SBW é baseado principalmente nos achados clínicos, sendo fundamental o reconhecimento precoce desta condição.
Palavras-chave: Síndrome de Beckwith Wiedemann, onfalocele, macroglossia
1 INTRODUÇÃO
A Síndrome de Beckwith-Wiedemann (SBW) é uma síndrome multissistêmica congênita caracterizada por hipoglicemia. Em 1964 Hans-Rudolf Wiedemann pediatra alemão descreveu um caso de um familiar que possuía onfalocele e macroglossia. Posteriormente o patologista pediátrico americano John Bruce Beckwith descreveu outros casos semelhantes e por este motivo foi batizada com o nome de síndrome de Beckwith-Wiedemann (SBW) (VIQUEIRA, E. B; RUBIO, R, U; PULIDO, J, C; 2014).
A incidência desta síndrome é de um caso para 13.500 nascidos vivos, a maioria dos casos ocorre de forma esporádica, mas a herança é complexa e inclui padrão autossômico dominante com uma expressividade variável e com a desregulação de genes na região cromossomal 11p15.5 (ARAÚJO JÚNIOR, E; et al, 2013). Não há predileção por raça ou idade, é maior em crianças geradas através de técnicas de reprodução assistida. A incidência é a mesma quando se leva em consideração ao sexo, com exceção de gêmeos monozigóticos onde há maior incidência em mulheres. Um em cada cinco casos morre devido a complicações da patologia (VIQUEIRA, E. B; RUBIO, R, U; PULIDO, J, C; 2014).
Vários mecanismos genéticos diferentes podem causar a síndrome. O mais comum desses mecanismos é um desequilíbrio na regulação de genes que estão envolvidos de forma variada no crescimento. Em cerca da metade, a SBW é causada por um distúrbio na metilação em IC2 com metilação reduzida do cromossomo 11 da mãe, que afeta a expressão dos genes CDKN1C, KCNQ1 e KCNQ1OT1 (CHOUFANI, S; SHUMAN; WEKSBERG, R; 2010). Na segunda causa mais comum (cerca de 20%), a criança recebeu um conjunto duplo do cromossomo 11 (todo ou parte) do pai: a UPD11 (dissomia uniparental do cromossomo 11) causa, entre outras coisas, uma superexpressão do gene IGF2, que estimula o crescimento. Em cerca de 85%, a síndrome ocorre esporadicamente. Isso significa que a síndrome não é herdada, aparecendo pela primeira vez na família. A probabilidade de os pais terem novamente um filho com a síndrome é então muito pequena (BARISIC, I; et al, 2018).
Na SBW, o mais importante é se a mutação foi herdada da mãe ou do pai. Quando a mutação é herdada da mãe, a probabilidade é de 50% de que seus filhos e filhas tenham a síndrome. Enquanto, se a mutação for herdada do pai, metade dos filhos terá a mutação, mas nenhum deles apresentará sintomas, porque apenas a cópia do gene da mãe está ativa. Se as filhas que têm a mutação tiverem filhos, a probabilidade é de 50% de que os filhos tenham a síndrome (CHOUFANI, S; SHUMAN; WEKSBERG, R; 2010).
O diagnóstico é realizado geralmente no período pós-natal através de sinais clínicos de macroglossia, onfalocele, gigantismo, visceromegalia, idade óssea avançada, displasia renal, nervos faciais e pregas na orelha (ARAÚJO JÚNIOR, E; et al, 2013). Como não há um consenso sobre os critérios clínicos diagnósticos, alguns autores sugerem diferentes critérios maiores e menores para o diagnóstico da SBW (Tabela 1) (NANCLARES, G. P. de; LAPUNZINA, P; 2015). Pacientes com esta síndrome tem uma predisposição a tumores embrionários aumentada em 7% em relação ao normal. Dentre eles os mais comuns são o hepatoblastoma e o tumor de Wilms (50%), os menos frequentes são o carcinoma adrenocortical, o neuroblastoma e rabdomiossarcoma, no caso de pacientes com hemi-hipertrofia existe um risco 30% maior de desenvolver tumores (VIQUEIRA, E. B; RUBIO, R, U; PULIDO, J, C; 2014).
Poderá ser feito o diagnóstico pré-natal desta síndrome, sendo este de grande importância para a evolução perinatal, para definir o tipo de parto, cuidado pediátrico principalmente decorrente da hipoglicemia neonatal, obstrução de vias aéreas superiores e falência cardíaca congestiva, além do risco de malignidade e acompanhamento com teste genético para membros da família. Pode ser realizado através de uma ultrassonografia bidimensional (US2D) por meio de dois critérios maiores ou um critério maior e dois menores (ARAÚJO JÚNIOR, E; et al, 2013).
As recomendações terapêuticas compreendem as principais questões clínicas em BWS, ou seja, monitoramento precoce de um risco aumentado de tumor, tratamento da macroglossia e defeitos da parede abdominal e intervenções terapêuticas para hipoglicemia (ELBRACHT, M; et al, 2018). Na região de cabeça e pescoço, pode apresentar várias alterações, as mais frequentes são proeminência occipital, achatamento do dorso nasal, hipoplasia maxilar e macroglossia. Em relação a esta última alteração, a macroglossia, pode causar, além do comprometimento estético, comprometimento funcional respiratório, com grande potencial de obstrução de vias aéreas superiores durante a infância ou adolescência. A fim de evitar episódios como este, alguns pacientes necessitarão realizar a glossectomia parcial (ASSIS; et al., 2012).
Apresentamos um caso de um recém-nascido, nascido de parto vaginal prematuramente e que possui manifestações clínicas que se enquadram nos critérios diagnósticos da SBW.
2 RELATO DE CASO
Paciente recém-nascido (RN), masculino, nascido de parto vaginal, em trânsito, durante transferência para o hospital da cidade de Campo Formoso-Bahia, às 5h do dia 15.03.23, com idade gestacional (IG) de 34 semanas e 6 dias. Dados do nascimento: comprimento: 50 cm, perímetro cefálico: 33 cm, peso ao nascer: 2.600 kg; APGAR: 8/9. Genitora fez 8 consultas de pré-natal, tinha história de hipertensão arterial sistêmica, em uso de metildopa. Relato de infecção do trato urinário de repetição durante a gravidez, com urocultura do 3º trimestre positiva para Proteus, sem controle de cura. Ultrassonografia (USG) do pré-natal evidenciando gastrosquise.
Durante o transporte, houve relato de vários episódios de hipoglicemia. Com aproximadamente 20 horas de vida, o RN foi admitido no Hospital Dom Malan em Petrolina – PE. Na admissão, estava em regular estado geral, dispneico, hidratado, corado, bem perfundido, ativo. A frequência respiratória era de 48 incursões por minuto e a saturação de oxigênio oscilava entre 85 e 90%. O abdome era depressível com presença de onfalocele; macroglossia; genitália masculina, com criptorquidia bilateral. O paciente foi internado, instalado suporte de oxigênio por pressão positiva contínua nas vias aéreas (CPAP), iniciada antibioticoterapia com ampicilina e gentamicina, solicitados exames laboratoriais e avaliação da cirurgia pediátrica. Ainda no primeiro dia de vida, foi realizada correção da onfalocele, procedimento sem intercorrências. No pós-operatório apresentou espasmos em membros associados a movimentos mastigatório e queda de saturação, aventada a suspeita de crise convulsiva, sendo realizada dose de ataque de fenobarbital, com melhora do quadro. RN fez uso de fenobarbital de manutenção durante o internamento, não apresentou novos episódios de crise convulsiva, sendo realizado desmame da medicação com boa tolerância.
Foi realizado parecer da cardiopediatria devido radiografia de tórax evidenciando aumento de área cardíaca, ecocardiograma com achados normais para a idade.
Durante o internamento o paciente apresentou vários episódios de cianose e apneia, com melhora após reposicionamento, aventando a hipótese de obstrução de vias aéreas como causa dessas intercorrências.
Paciente evoluiu com melhora clínica progressiva, em aleitamento materno complementado com fórmula infantil modificada, recebendo alta hospitalar com 21 dias de vida e encaminhamento para acompanhamento ambulatorial.
A presença de hipoglicemia, criptorquidia, macroglossia, com grave tendência à obstrução de vias aéreas superiores, aliados à presença de onfalocele, preenchem os critérios diagnósticos da Síndrome de Beckiwith-Wiedemann.
Na tabela 1 contém um resumo dos exames realizados e seus resultados pelo paciente.
Tabela 1
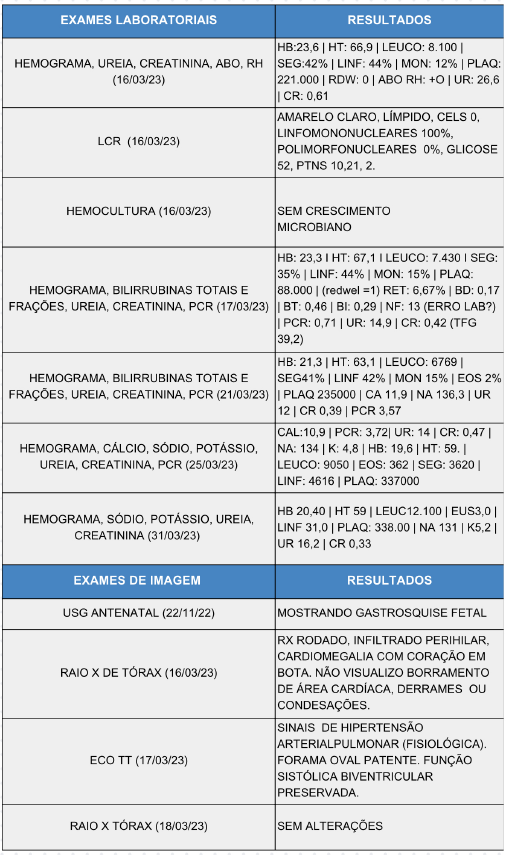
3 DISCUSSÃO
Um grande estudo sobre a SBW feito em 16 países europeus no período de 1990 a 2015, identificou 371 possíveis casos, houve 15 interrupções da gestação (4,0%) após detecção ultrassonográfica pré-natal de anomalias graves, como por exemplo onfalocele; 10 óbitos fetais (2,7%); e 346 nascidos vivos (93,3%). Dos nascidos vivos, 12 morreram na primeira semana de vida (3,6%), e destes, 11 eram prematuros (91,6%). (Barisic et al; 2018).
O diagnóstico desta condição, em geral, é realizado no período pós-natal por meio dos achados de macroglossia, onfalocele, gigantismo, visceromegalia, idade óssea avançada, displasia renal, nevos faciais e pregas na orelha. O diagnóstico pré-natal da SBW pela ultrassonografia bidimensional pode ser realizado por meio de dois critérios maiores ou um critério maior e dois menores. Os critérios maiores incluem macroglossia, macrossomia (estimativa de peso acima do percentil 90 para idade gestacional) e um defeito da parede abdominal. Os critérios menores incluem polidrâmnio, nefromegalia, displasia renal e citomegalia adrenal confirmada por diagnóstico patológico. A ultrassonografia tridimensional no modo de renderização permite melhor avaliação da superfície fetal, contribuindo para melhor entendimento das malformações pelos pais. (ARAÚJO JÚNIOR et al; 2013).
Tabela 2 Critérios diagnósticos para a SBW

Fonte: Fonte adaptada de Critérios diagnósticos segundo Elliot e Cols.
Um outro estudo realizado no Hospital Infantil da Filadélfia, em 2014, com 344 pacientes com espectro de Beckwith-Wiedemann. Dentre eles, 207 (64,3%) tiveram como características clínicas designadas como clássicas (macroglossia, onfalocele, crescimento excessivo); 60 (18,6%) com supercrescimento lateralizado isolado; e 55 (17,1%) designados como atípicos, que são os pacientes com defeito molecular (DUFFY et al; 2019).
Segundo Duffy et al; 2019, a característica clínica mais prevalente foi a de crescimento excessivo lateralizado (74,2%), seguido de macroglossia (66,5%), defeito na parede abdominal (65,9%), pregas/fossas auriculares (58,3%), hipoglicemia (53,3%), prematuridade (43,6%), hérnia umbilical (34%), criptorquidia (30,1%), onfalocele (19,6%), tumoração (14,5%).
O diagnóstico diferencial da SBW inclui diabetes materno, outras síndromes de crescimento excessivo como Simpson-Golabi-Behmel, Perlman, Sotos, Costello, espectro de supercrescimento relacionado à PIK3CA e algumas doenças raras como síndrome tumoral de hamartoma PTEN ou mosaicismo de trissomia do cromossomo 8. Através de investigações adequadas e clínica bem-feita, essas condições podem ser diferenciadas sem dificuldades (WEKSBERG et al; 2010; BRIOUDE et al; 2018).
É muito importante o diagnóstico em pacientes com menor expressividade fenotípica devido ao alto risco associado de desenvolvimento de tumores embrionários e de hipoglicemias severas. O acompanhamento do paciente com a síndrome consiste em cirurgia de correção de defeitos da parede abdominal, triagem de hipoglicemia e tratamento, se necessário, triagem tumoral e somatometria. Alguns pacientes podem precisar de cirurgia redutora da língua por dificuldade na ingestão ou obstrução das vias aéreas. A possibilidade de apneia do sono tem de ser monitorada. Geralmente esses indivíduos precisam do apoio da equipe de fonoaudiologia e odontologia. (BULLER VIQUEIRA et al; 2014; CIELO et al; 2019).
Como citado anteriormente no relato de caso, o paciente em questão apresenta macroglossia, onfalocele, hipoglicemia, criptorquidia, e que não houve o diagnóstico no período pré-natal, por meio de ultrassonografia. Em síntese, acreditamos que a ultrassonografia possa contribuir para uma melhor avaliação de algumas malformações no pré-natal como a SBW, possibilitando o diagnóstico precoce, prevenção de complicações e uma melhor compreensão da doença pelos pais, bem como pela equipe multidisciplinar. (ARAÚJO JÚNIOR et al; 2013).
O SBW tem uma baixa incidência que pode ser tendenciosa pela falta de diagnóstico em casos de menor expressividade clínica. O paciente portador da síndrome necessita de acompanhamento multidisciplinar contínuo, devido ao risco de complicações associadas às malformações e risco de neoplasias malignas (BULLER VIQUEIRA E et al, 2014)
4 CONCLUSÃO
O diagnóstico de pacientes com SBW é baseado nos achados clínicos, sendo fundamental o reconhecimento precoce, pelo risco maior de surgimento de tumores na infância e complicações ainda no período neonatal. O prognóstico dos nascidos vivos é favorável, com alta sobrevida no primeiro ano, cerca de 9,1% dos pacientes morreram nesse período (BARISIC et al; 2018). Já no espectro grave, os doentes correm risco de morte prematura devido a complicações decorrentes da hipoglicemia, prematuridade, cardiomiopatia, macroglossia ou tumores. Em doentes que sobrevivem à infância, o prognóstico é geralmente bom (NANCLARES, G. P. de; LAPUNZINA, P; 2015).
O controle da síndrome envolve estratégias típicas de suporte médico e cirúrgico. É necessária a vigilância tumoral, que deverá ser iniciada se o BWS for suspeitado e/ou diagnosticado, além do aconselhamento genético que se faz extremamente recomendado, a estimativa do risco de recorrência e testes genéticos em cascata devem contar com a história familiar e subgrupo molecular do membro da família afetado (NANCLARES, G. P. de; LAPUNZINA, P; 2015).
Faz-se necessário que a criança com SBW receba um acompanhamento interdisciplinar, envolvendo Fonoaudiologia, Ortodontia, Cirurgia Bucomaxilofacial, dentre outros profissionais, desde o seu nascimento para um efetivo manejo da síndrome, para que possa resultar na adequação das funções orofaciais e corporais, tendo assim um desenvolvimento adequado evitando limitações.
REFERÊNCIAS
ARAÚJO JUNIOR, E. A.; SIMIONI, C.; NARDOZZ, L. M. M.; MORON, A. F. Diagnóstico pré-natal da síndrome de Beckwith-Wiedemann pela ultrassonografia bidimensional e tridimensional. Relato De Caso. R Radiol. Bras. 46 (6). Nov-Dec. 2013. https://doi.org/10.1590/S0100-39842013000600012.
ASSIS, G. M. de; CAMPOS, G. B. P.; SILVA, J. S. P. da; GERMANO, A. R. Glossectomia parcial em paciente com síndrome de Beckwith-Wiedemann – Caso Clínico. Revista Extensão & Sociedade, [S. l.], v. 1, n. 4, 2012. Disponível em: https://periodicos.ufrn.br/extensaoesociedade/article/view/1636. Acesso em: 22 maio.2023.
BARISIC, I; BOBAN, L; AKHMEDZHANOVA, D; BERGMAN, J. E. H; CAVEROCARBONELL, C; GRINFELDE, I. Síndrome de Beckwith Wiedemann: Um estudo de base populacional sobre prevalência, diagnóstico pré-natal, anomalias associadas e sobrevivência na Europa. Eur J Med Gen; 61: 499-507, 2018.
Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, Boonen SE, Cole T,
Baker R, Bertoletti M, Cocchi G, Coze C, De Pellegrin M, Hussain K, Ibrahim A, Kilby
MD, Krajewska-Walasek M, Kratz CP, Ladusans EJ, Lapunzina P, Le Bouc Y, Maas
SM, Macdonald F, Õunap K, Peruzzi L, Rossignol S, Russo S, Shipster C, Skórka A,
Tatton-Brown K, Tenorio J, Tortora C, Grønskov K, Netchine I, Hennekam RC, Prawitt D, Tümer Z, Eggermann T, Mackay DJG, Riccio A, Maher ER. Documento de consenso de especialistas: Diagnóstico clínico e molecular, rastreamento e manejo da síndrome de Beckwith-Wiedemann: uma declaração de consenso internacional. Nat Rev Endocrinol. 2018 Abr;14(4):229-249. DOI: 10.1038/nrendo.2017.166. EPub 2018 29 de janeiro. PMID: 29377879; PMCID: PMC6022848.
BULLER VIQUEIRA, Eva; UREBA RUBIO, Rosalía; CABELLO PULIDO, Juana. Síndrome de Beckwith-Wiedemann. Rev Clin Med Fam, Barcelona , v. 7, n. 1, p. 6668, feb. 2014.
CIELO, C. M; DUFFY, K. A; TAYLOR, J. A; MARCUS, C. L; KALISH, J. M. Apneia obstrutiva do sono em crianças com síndrome de Beckwith-Wiedemann. 2019 Mar 15;15(3):375-381. DOI: 10.5664/jcsm.7656. PMID: 30853040; PMCID:PMC6411187.
CHOUFANI, S; SHUMAN; WEKSBERG, R. Síndrome de Beckwith-Wiedemann. Am J Med Genet; 154C: 343-354; 2010.
DeBAUN, M.R; TUCKER, M.A. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. Pediatr; 132:398400;1998.
DUFFY, K. A; CIELO, C. M; COHEN, J. L; GONZALEZ-GANDOLFI, C. X; GRIFF,
J.R; HATHAWAY, E. R; KUPA, J; TAYLOR, J.A; WANG, K. H; GANGULY, A;
DEARDORF, M. A; KALISH, J. M. Caracterização do espectro de BeckwithWiedemann: diagnóstico e conduta. Sou J med genet C semin med genet. 2019 Dez;181(4):693-708. DOI: 10.1002/ajmg.c.31740. EPub 2019 30 de agosto. PMID: 31469230; PMCID: PMC7959855.
ELBRACHT, M; PRAWITT, D; NEMETSCHEK, R; KRATZ, C; EGGERMANN, T. Beckwith-Wiedemann-Syndrom (BWS): Aktueller Stand der Diagnostik und des klinischen Managements [Beckwith-Wiedemann Syndrome (BWS) Current Status of Diagnosis and Clinical Management: Summary of the First International Consensus Statement]. Klin Padiatr. 2018 Apr;230(3):151-159. German. doi: 10.1055/a-05919479. Epub 2018 Apr 16. PMID: 29660755.
ELLIOTT, M; BAYLY, R; COLE, T; TEMPLE, I. K; MAHER, E. R. Clinical features and natural history of Beckwith-Wiedemann syndrome: presentation of 74 new cases. Clin Genet. 46:168-74; 1994.
NANCLARES, G. P. de; LAPUNZINA, P. Enfermedades de Impronta: Guías de buena práctica clínica. Cap.4. 260p. CIBERER, 2015.
VIQUEIRA, E. B; RUBIO, R, U; PULIDO, J, C. Síndrome de Beckwith-Wiedemann. Rev Clin Med Fam , Barcelona, v. 7, n. 1, pág. 66-68, fev. 2014. Disponível em <http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1699-695X2014000100012&lng=es&nrm=iso>. acessado em 19 de maio de 2023. https://dx.doi.org/10.4321/S1699-695X2014000100012.
WEKSBERG, R; SHUMAN, C; BECKWITH, J. B. Beckwith-Wiedemann syndrome. Eur J Hum Genet; 18:8-14; 2010.
ANEXO A
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO
RELATO DE CASO CLÍNICO
O(a) Sr.(a) está sendo consultado(a) no sentido de autorizar a utilização de dados clínicos, laboratoriais, imagens fotográficas e lâminas histológicas de seu caso clínico que se encontram em sua ficha de prontuário médico) para finalidades científicas (apresentação em congressos ou publicação do caso em revista científica) sob a forma de relato de caso clínico.
Nosso objetivo será o de discutir as características de sua doença em meio científico, em função das particularidades de apresentação de sua doença, metodologia de diagnóstico e tratamento utilizado. Não há riscos estabelecidos neste procedimento e, adicionalmente, a discussão do caso servirá como ferramenta para o estudo acadêmico e contribuição à ciência. Para participar deste estudo, o(a) Sr.(a) não terá nenhum custo, nem receberá qualquer vantagem financeira.
A sua autorização é voluntária e a recusa em autorizar não acarretará qualquer penalidade ou modificação na forma em que é atendido(a) pelos médicos assistentes e pesquisadores. Os pesquisadores irão tratar a sua identidade com padrões profissionais de sigilo, atendendo à legislação brasileira (Resolução Nº. 466/12 do Conselho Nacional de Saúde), utilizando as informações somente para fins acadêmicos e científicos. O relato do caso estará à sua disposição quando finalizado. Seu nome ou o material que indique sua participação não será liberado sem a sua permissão. O(a) Sr.(a) não será identificado(a) em nenhuma publicação. Este termo de consentimento encontra-se impresso em duas vias, sendo que uma cópia será arquivada pelo pesquisador responsável, e a outra será fornecida ao(à) Sr.(a).
Eu, Elisangela da Silva Vieira fui informada a respeito do objetivo deste estudo, de maneira clara e detalhada e esclareci minhas dúvidas. Sei que a qualquer momento poderei solicitar novas informações. Declaro que autorizo a utilização de dados clínico-laboratoriais de meu caso. Recebi uma cópia deste termo de consentimento livre e esclarecido e me foi dada a oportunidade de ler e esclarecer as minhas dúvidas.
Campo Formoso-BA, 15 de maio de 2023.

*Acadêmicos de Medicina da Faculdade Estácio de Juazeiro – BA
**Médica Pediatra e Professora do Curso de Medicina da Faculdade Estácio de Juazeiro – BA