IVACAFTOR ASSOCIATED WITH OTHER DRUGS FOR THE TREATMENT OF CYSTIC FIBROSIS
REGISTRO DOI: 10.5281/zenodo.7742248
Flávio Henrique Cesar Drummond¹
Maria Fernanda Sanchez Perez¹
Thaís Lamounier Santos¹
Vitor Ferrari Cossi¹
RESUMO
INTRODUÇÃO: A Fibrose Cística é uma doença autossômica recessiva, causada por uma mutação no gene CFRT (cystic fibrosis transmembrane conductance regulator), responsável pela codificação de proteínas transmembranas para o íon cloreto. OBJETIVO: Analisar estudos envolvendo o uso de ivafactor em pacientes com fibrose cística, analisando o mecanismo de ação do medicamento de acordo com as dosagens e as possíveis mudanças fenotípicas provocadas nas demasiadas pesquisas. MÉTODOS: Revisão sistemática e integrativa de estudos randomizados e de coorte transversal, usando as bases de dados Scielo, Pubmed, Dynamed e as palavras chaves “Cystic Fibrosis”, “CFRT” e “Ivafactor” com publicações entre os anos de 2011 a 2021. RESULTADOS: Os estudos apresentaram melhorias significativas das funções respiratórias nos pacientes que receberam ivafactor quando comparados aqueles que receberam o placebo. Ainda, as pesquisas demonstraram que a combinação tripla de Tezacaftor / Ivacaftor/ Elexacaftor, além de resultados positivos benéficos para a manifestação clínica do gene CFRT, houve poucos eventos adversos graves (4%). CONCLUSÃO: A combinação Tripla de Elexacaftor, Tezacaftor e Ivacaftor se mostrou significativa na modulação do gene CFTR e com boa eficácia terapêutica, provocando, também, melhorias nutricionais. Eventos adversos da combinação de Elexacaftor, Tezacaftor e Ivacaftor foram leves e de ágil resolução. A associação de Ivacaftor e Tezacaftor teve um prognóstico geral melhor quando comparados aqueles que receberam monoterapia de Tezacaftor. O uso exclusivo de Ivacaftor reduziu as chances de exacerbação pulmonar, resultou no aumento do volume expiratório forçado e apresentou aumento da atividade proteica do CFTR após um mês de uso.
PALAVRAS-CHAVE: Fibrose cística. Ivafactor. CFRT
ABSTRACT
BACKGROUND: Cystic fibrosis is an autosomal recessive disease caused by a mutation in CFTR (cystic fibrosis transmembrane conductance regulator) gene, responsible for the expression of chloride transmembrane protein. OBJECTIVE: analyze studies involving the Ivacaftor`s use in patients with cystic fibrosis, observing its mechanism of action, efficacy, and safety. METHODS: Systematic and integrative review of the existing bibliography using databases like Scielo, Pubmed, and Dynamed and using the keywords “Cystic fibrosis”, “CFTR” and “Ivacaftor”, in addition to articles published between 2015 and 2021. RESULTS: The studies show significant improvement in respiratory functions in patients that received the Ivacaftor treatment in comparison to those who received a placebo. Besides, the researches reveal that the association of Ivacaftor with other drugs as Tezacaftor, Lumacaftor, and Elexacaftor contributed to improve the effectiveness of the treatment. CONCLUSION: The triple combination of Elaxacaftor Tezacaftor and Ivacaftor has shown a significant outcome on CFTR gene modulation and a great therapeutic efficacy, proving, as well, nutritional improvement. Adverse events of Elexacaftor, Tezacaftor, and Ivacaftor combination were of mild severity and fast resolution. The Ivacaftor association with Tezacaftor has had a greater prognosis in comparison to the Tezacaftor monotherapy. The exclusive use of Ivacaftor reduced the pulmonary exacerbation, resulted in an increase of forced expiratory volume, and showed an improvement of CFTR`s proteic activity after one month of use.
KEYWORDS: Cystic Fibrosis. Ivacaftor. CFTR
1 INTRODUÇÃO SOBRE A FIBROSE CÍSTICA: GENÉTICA, EPIDEMIOLOGIA E CLÍNICA
1.1 Introdução genética
A fibrose cística (FC) é uma doença de herança autossômica recessiva no braço longo cromossomo 7 (7q31.2), identificada pela primeira vez em 19894 , que afeta principalmente o pâncreas, os pulmões, os intestinos e o fígado. O gene da fibrose Cística codifica 1480 aminoácidos¹ e, atualmente, tem-se um conhecimento, aproximado, de mais de duas mil mutações do gene CFRT. A mutação mais comum, logo, a mais presente em estudos clínicos, é a mutação do tipo F508del.4 O produto final do gene CFRT é uma proteína transmembrana localizada na superfície apical do tecido epitelial, queratinizado ou não queratinizado, que atua no transporte de íons cloreto. A disfunção dessa proteína pode variar em até seis níveis, desde nenhuma produção proteica, até a presença de proteína instável na membrana celular. A perda funcional da proteína ou sua ausência provoca, no tecido epitelial, impedimento da reabsorção de cloro da superfície epitelial para o meio extracelular, aumentando a concentração do íon Cl- Por outro lado, no pulmão, ocorre processo inverso, com fluxo aumentando de íons da superfície epitelial para o interstício.
1.2 Introdução Epidemiológica
No Brasil, estima-se uma incidência de 1 a cada 7.358 nascimentos, com maior estimativa no Rio Grande do Sul, e a menor em São Paulo². Por outro lado, no mundo, estima-se uma prevalência de 70.000 casos da doença em crianças, sendo o maior percentual de mutações do tipo F508del ³. A doença se faz mais presente na população branca de origem caucasiana, como europeus, norte americanos e australianos ³.
1.3 Introdução clínica
No Brasil, a partir de 2001, a triagem neonatal passou a incluir a FC no grupo das doenças diagnosticadas por meio do “teste do pezinho”, sendo este obrigatório e gratuito para qualquer recém-nascido. A dosagem do tripsinogênio imunorreativo (IRT), com medida positiva, é sugestiva para FC. O exame laboratorial deve ser realizado uma segunda vez, haja vista a probabilidade de resultados falsos positivos. Após os dois exames positivos, o teste do suor, padrão ouro para o diagnóstico da FC, deve ser requisitado. Esse teste afere a quantidade de íons cloreto secretados durante a transpiração do neonato, tendo valores superiores à 60mmol/L indicativo da doença. Por fim, a avaliação genética vai confirmar se a criança é portadora da mutação do gene CFRT.
As manifestações clínicas gerais, ou seja, que ocorrem em qualquer idade, envolvem inchaço das falanges distais, conhecido como baqueteamento digital, alcalose metabólica hipoclorêmica devido, por exemplo, ao aumento da concentração de íons cloreto no suor, provocando “sabor salgado”. Além destes, o sinal clínico clássico é a tosse com a produção elevada de muco expectorante, provocando-se, quando em excesso, dificuldade para dormir e alimentação inadequada do lactente portador. As variações mutacionais da FC podem interferir nas manifestações clínicas do paciente 4. Os níveis funcionais da mutação CFTR podem provocar desde insuficiência pancreática, em 85% dos casos 14, até pacientes assintomáticos. Pessoas que cursam com a sintomatologia mais branda da doença ou assintomáticos são, muitas vezes, diagnosticadas pelo grau de parentesco próximo com algum portador da Fibrose Cística.
As terapias medicamentosas, utilizando o ivafactor, devem ser, cuidadosamente, analisadas para o tipo de mutação do gene CFRT. Para tanto, cada tipo de mutação, atrelada à idade do paciente, necessita-se utilizar uma dosagem medicamentosa específica e uma combinação com outros fármacos, como tezacaftor e lumacaftor. Outrossim, os quadros mais graves da doença podem evoluir para quadros de transplantes pulmonares e a necessidade de sonda enteral.
2 RESULTADOS
Tabela 1:
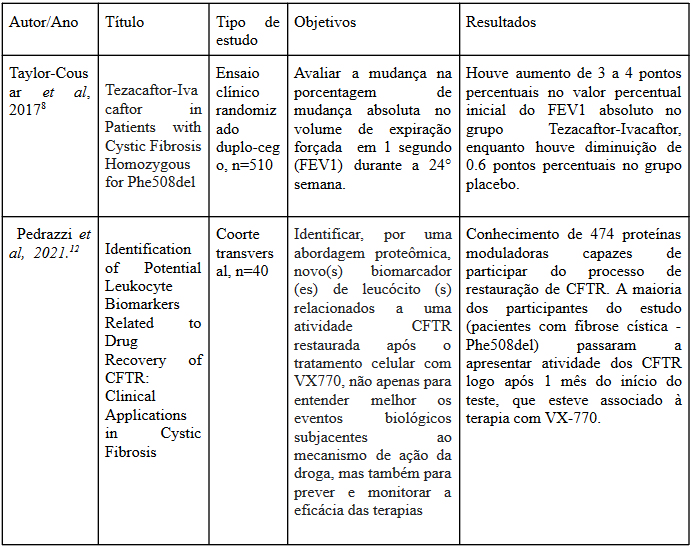
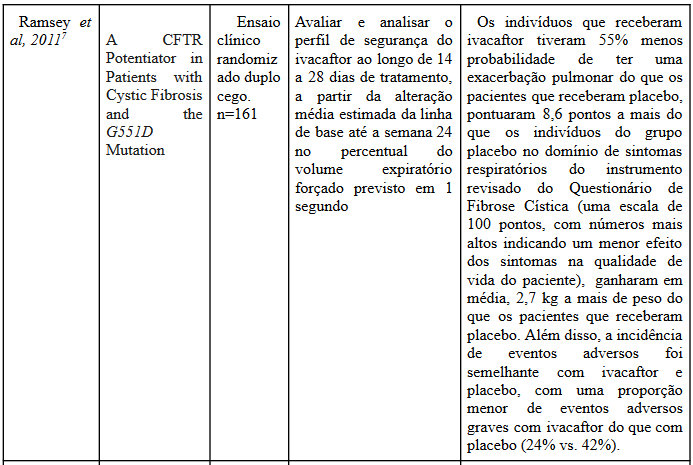

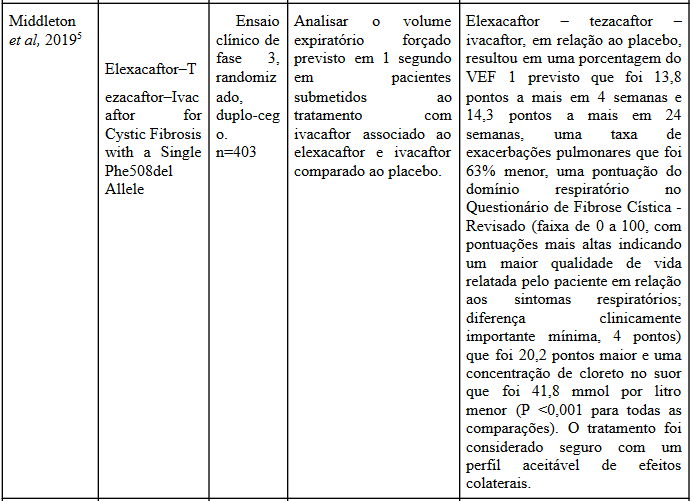
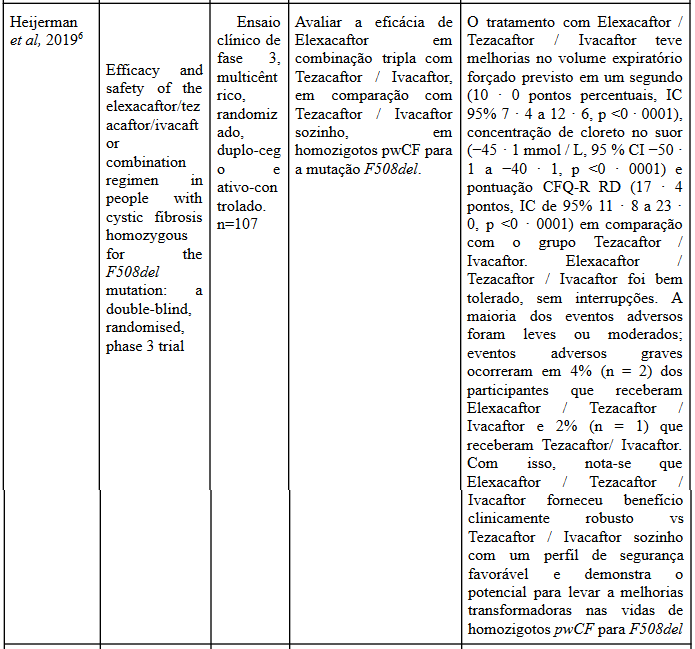
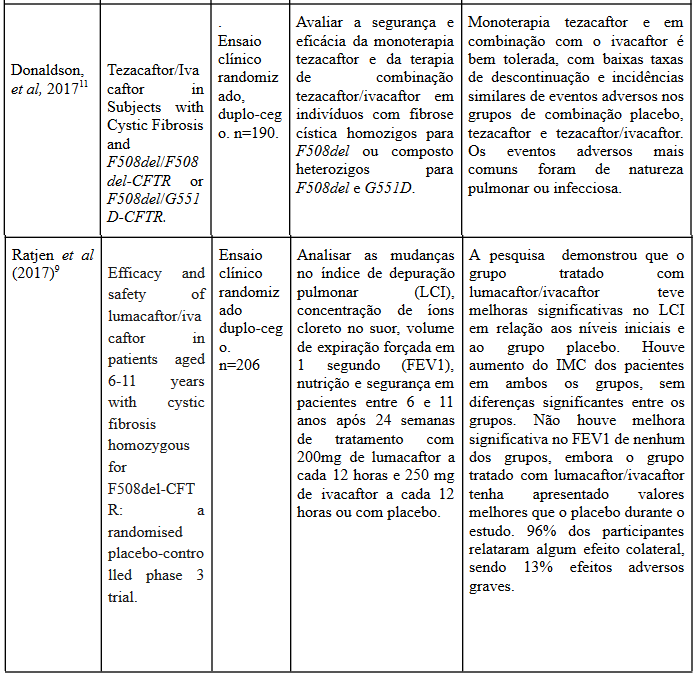
2 DISCUSSÃO
2.1 O uso do Ivacaftor e combinação com outras drogas
O medicamento estudado auxilia no tratamento da fibrose cística ao potencializar a ação da proteína CFTR em pacientes em que há defeito na produção da mesma de forma a prejudicar o seu funcionamento normal. Assim, o fármaco em questão aumenta a capacidade de transportar íons cloreto da CFTR defeituosa de forma a estimular um funcionamento mais próximo do ideal.
Conforme o apresentado nos resultados das pesquisas feitas para a elaboração deste artigo, o Ivacaftor é na maioria das vezes administrado a pacientes com fibrose cística associado a outros fármacos com ações diferentes relacionadas ao CFTR para melhorar a eficácia do tratamento para cada tipo de mutação na proteína.
A associação do Ivacaftor ao fármaco Lumacaftor leva a uma melhora no quadro de pacientes que apresentam a mutação F508del, uma vez que o Lumacaftor corrige e melhora a conformação da proteína de forma a permitir maior processamento e tráfego da CFTR madura para a membrana plasmática das células, enquanto o Ivacaftor potencializa a abertura do canal para transporte dos íons cloreto.
Outra combinação de remédios conhecida e estudada é a associação entre Elexacaftor, Tezacaftor e Ivacaftor. O Elexacaftor e o Tezacaftor têm funções semelhantes à do
Lumacaftor, pois se combinam para facilitar o processamento e o transporte da proteína CFTR à superfície da célula enquanto o Ivacaftor potencializa a ação da proteína. O
Tezacaftor também é utilizado em associação ao Ivacaftor sem a presença do Elexacaftor.
2.2 Tratamento com Ivacaftor isolado
O artigo de Ramnsey et al (2011)7, desenvolve um estudo do tratamento com ivacaftor isolado, obtendo resultados um aumento na linha de base do volume expiratório forçado, enquanto o grupo placebo houve uma diminuição desta linha. Assim, o medicamento isolado refletiu num incremento significativo no volume expiratório forçado, e este efeito considerável foi mantido ao longo do estudo, com duração de 48 semanas.
Além disso, os indivíduos tratados com ivacaftor, em comparação com aqueles que receberam placebo, indicaram uma redução nos sintomas respiratórios, evidenciando um aumento nas pontuações do domínio respiratório do CFQ-R. Em seguida, pacientes tratados com o ivacaftor, no decorrer do estudo, adquiriram 3,1 kg, em comparação com o placebo, que foi de somente 0,4kg (efeito do tratamento, 2,7 kg; P <0,001).
Outrossim, analisando o estudo de coorte de Pedrazzi et al, a atividade da proteína CFTR foi aumentada em indivíduos tratados com ivacaftor após um mês de estudos.
Portanto, com os resultados dos estudos, conclui-se a existência de benefícios ao tratamento da FC ao se utilizar o ivacaftor isolado, que tem efeito na atividade do CFTR, logo, aumentando a saída de íons cloreto e água para fora das células, o que diminui a viscosidade das secreções pulmonares levando a uma melhora nos sintomas respiratórios, com isso, o quadro clínico dos pacientes com fibrose cística pode ser atenuado.
Destarte, voltando ao estudo de Ramnsey et al7, a incidência de eventos adversos nos dois grupos estudados foi semelhante. Porém, o grupo tratado com ivacaftor tiveram menor incidência de eventos adversos que levaram a descontinuação do estudo, 1% vs 5% no placebo.Tais eventos no grupo placebo foram; aumento dos níveis das enzimas hepáticas, bloqueio atrioventricular, ataque de pânico e insuficiência respiratória, enquanto no grupo de ivacaftor foi aumento dos níveis das enzimas hepáticas somente.
Deste modo, eventos adversos que ocorreram com mais frequência no grupo ivacaftor foram cefaléia, infecção do trato respiratório superior, congestão nasal, erupção cutânea e tontura; nenhum desses eventos foi considerado sério ou levou à descontinuação do medicamento em estudo.
E por fim, houve uma taxa mais baixa de eventos adversos graves no grupo de ivacaftor (24%) que no placebo (42%)7, não ocorrendo nenhuma morte no estudo. Com isso, consequentemente, após a pesquisa, pode-se afirmar que o tratamento com ivacaftor não foi associado a um menor risco de segurança do que o observado com o placebo, tendo até mesmo eventos adversos graves menos comuns.
Em síntese, os artigos expõem um alternativa de tratamento projetado para melhorar a função da proteína CFTR como um meio de abordar a causa subjacente da fibrose cística e começar a cumprir a promessa anunciada com a descoberta do gene CFTR, deste modo a utilização de ivacaftor de forma isolada é potencialmente benéfico para o tratamento da fibrose cística.
2.3 Tratamento com Ivacaftor associado ao Lumacaftor
Tanto o artigo de Wainwright et al (2015)10 quanto o artigo de Ratjen et al (2017)9 visualizaram, a partir de seus estudos, que houve uma melhora absoluta média entre os pacientes tratados com lumacaftor-ivacaftor, desde a linha de base até a semana 24. Relataram aumentos significativos no IMC médio, ao longo do período de 24 semanas, dos grupos de dose lumacaftor-ivacaftor, se comparados com grupos placebos. A diferença em relação à mudança absoluta de tratamento versus placebo no IMC foi de 0,24 a 0,28 (P<0,001), o que representa uma melhora de aproximadamente 1% com a adesão do lumacaftor-ivacaftor. Ademais, ambos autores concordam que em grupos de dose lumacaftor-ivacaftor as pontuações do domínio respiratório do CFQ-R foram melhoradas em relação à linha de base até a semana 24.
Os pesquisadores destacaram, ainda, que houve elevações nos níveis de aminotransferase alanina ou aspartato aminotransferase para mais de 3 vezes o limite superior da faixa normal em cerca de 13% dos pacientes do grupo lumacaftor- ivacaftor. Ratjen et al9 assinala que a taxa de elevações de aminotransferase em pacientes mais jovens foi consistente com os resultados esperados em pacientes pediátricos com fibrose cística, que apresentam uma alta taxa de fundo de anormalidades de aminotransferase 31. Já Wainwright et al10 lembra que eventos adversos graves relacionados à função hepática anormal não foram observados no grupo placebo e foram relatados para sete pacientes nos grupos lumacaftor-ivacaftor.
Conforme Ratjen e colaboradores9, a maioria dos eventos adversos foram respiratórios ou infecciosos e foram relatados tanto no grupo lumacaftor-ivacaftor, quanto no grupo placebo.
Foi observado, nesses estudos, que eventos adversos (tosse produtiva, congestão nasal, dor orofaríngea e dor abdominal) foram mais frequentes no grupo de tratamento com lumacaftor-ivacaftor do que com placebo. Todavia, consoante Wainwright e colaboradores10, tais eventos foram relatados em maior número no grupo placebo, sendo que o padrão desses foi semelhante entre os grupos placebo e lumacaftor-ivacaftor, de modo que, nos grupos lumacaftor-ivacaftor, a maioria era de gravidade leve a moderada e incluía dispneia e aperto no peito.
Wainwright10 e equipe apontaram que menores taxas de exacerbações pulmonares foram observadas nos grupos de dose lumacaftor-ivacaftor, e em menor frequência que no grupo placebo. Como resultado, se observou que durante a semana 24, a proporção de pacientes que permaneceram livres de exacerbações foi maior nos grupos lumacaftor-ivacaftor do que no grupo placebo, ao passo que o risco de ter uma exacerbação foi significativamente menor naqueles grupos. Já Ratjen9 e autores afirmaram que a alteração absoluta da linha de base de FEV até a semana 24 não foi significativa em nenhum dos grupos de tratamento. Esse dado difere do encontrado no artigo de Wainwright10, o qual aponta que o percentual de pacientes que apresentaram melhora relativa no percentual de FEV de 5% ou mais foi maior nos grupos lumacaftor-ivacaftor do que no grupo placebo.
2.4 Tratamento com Ivacaftor associado ao Tezacaftor
Os artigos estudados que apresentam o tratamento associado de Ivacaftor e Tezacaftor têm resultados semelhantes com um prognóstico geral melhor para os pacientes que receberam o tratamento combinado em relação ao grupo placebo e ao grupo que recebeu monoterapia de Tezacaftor.
Taylor-Cousar et al (2017)8 descrevem principalmente os valores de FEV1, concentração de cloreto no suor, CFQ-R RD e IMC para indicar e medir a progressão do quadro em cada paciente, obtendo uma melhora significativa em quase todas as variáveis indicadas para o grupo que recebeu tratamento, exceto no IMC, em que a diferença dos aumentos não foi tão relevante entre os grupos.
De forma semelhante, Donaldson et al (2017)11 traz uma análise da progressão da doença em três grupos diferentes, sendo um placebo, um com monoterapia de Tezacaftor e um com associação de Tezacaftor e Ivacaftor em diferentes dosagens, possibilitando não só analisar a eficácia do tratamento mas comparar tipos diferentes terapia.
Nessa segunda pesquisa foram achados que os valores de FEV1 nos grupos que receberam a monoterapia e terapia combinada se apresentaram significativamente maiores e os valores de concentração de cloreto no suor significativamente menores em ambos os grupos, principalmente nos pacientes que receberam terapia combinada.
Assim, percebe-se que os artigos estão em concordância a respeito da eficácia dos tratamentos com Ivacaftor e Tezacaftor uma vez que ambos obtiveram um bom desfecho.
No que tange à segurança do tratamento, os dois artigos indicam que não há aumento relevante no número de eventos adversos entre os grupos placebo e que receberam tratamento, sendo em sua maioria eventos de baixa gravidade. Segundo Taylor-Cousar et al
(2017)8, dos 78 casos de eventos adversos graves apenas 31 vieram de pacientes que receberam tratamento e 47 dos grupos placebo, levando a entender que o uso dos medicamentos em questão não só não apresenta risco como diminui a incidência de eventos adversos nos pacientes.
2.5 Tratamento com Ivacaftor associado ao Elexacaftor e Tezacaftor
A partir da análise dos estudos de Middleton et al (2019)5 e Heijerman et al (2019)6 o tratamento com elexacaftor – tezacaftor – ivacaftor resultou em melhorias expressivas significativa no desfecho primário da mudança absoluta em porcentagem do VEF, levando a uma melhora rápida no ppFEV, também houve um aumento na concentração do cloreto no suor. Quando o tratamento foi comparado com TEZ / IVA somente, a diferença na pontuação CFQ-R RD foi considerável.
Além disso, a combinação desses três medicamentos resultaram em menor taxa de exacerbações pulmonares do que o placebo (razão de taxas, 0,37; intervalo de confiança de 95%, 0,25 a 0,55; P <0,001)5,6, fator que também pode ser extrapolado para taxa de exacerbações que levaram à hospitalização ou que foram tratadas com antibióticos intravenosos, com isso, uma taxa menor de pacientes que tomaram a combinação do que no grupo placebo permaneceram livres de exacerbação. Ademais, o tratamento com ELX / TEZ / IVA gerou um aumento no IMC em comparação com o TEZ/IVA5,6.
Eventos adversos ocorreram em 10% dos pacientes em ambos os grupos, sendo que a maioria dos pacientes em tratamento de Elexacaftor – tezacaftor – ivacaftor, majoritariamente tiveram eventos adversos leves de rápida resolução e apenas um pequena porcentagem teve algum evento adverso grave5.
Portanto, de acordo, com os estudos analisados a terapia moduladora de CFTR de combinação tripla, Elexacaftor – tezacaftor – ivacaftor, resultou em melhora significativa em várias medidas importantes e também modulando fortemente o CFTR, sendo então altamente eficaz para terapêutica da doença, gerando até mesmo benefícios nutricionais. Em conclusão, foi demonstrado a eficácia e a segurança do tratamento ELX/TEZ/IVA, que possui grande potencial terapêutico, podendo gerar um imenso impacto no quadro do paciente com FC.
3 CONCLUSÃO
A partir dessa revisão integrativa, percebe-se o avanço no tratamento da Fibrose Cística, com melhorias significativas na vida dos pacientes portadores da doença. Os fármacos utilizados no tratamento se mostraram de grande valia na amenização dos sintomas e, ainda, geraram benefícios indiretos à saúde, como ganho de peso devido à ampliação da absorção nutricional em virtude do aumento da atividade da proteína CFTR.
Com tudo, ainda são poucos e recentes os relatos na literatura, necessitando-se de mais estudos acerca de terapias medicamentosas para a Fibrose Cística. Em decorrência de pacientes, mesmo que poucos, terem tido reações graves ao fármacos, deve haver a manutenção das pesquisas para descobrir as causas das reações adversas para compreendê-las e, ainda, evitá-las.
REFERÊNCIA BIBLIOGRÁFICA
1- RIORDAN; ROMMENS, J.; KEREM, B; ALON, N; ROZMAHEL, R; GRZELCZAK, Z; ZIELENSKI, J; LOK, S; PLAVSIC, N; CHOU, J.. Identification of the cystic fibrosis gene: cloning and characterization of complementary dna. Science, [S.L.], v. 245, n. 4922, p. 1066-1073, 8 set. 1989. American Association for the Advancement of Science (AAAS). http://dx.doi.org/10.1126/science.2475911. Acesso em: 02 de junho de 2021.
2- FIRMIDA, Mônica de Cássia; LOPES, Agnaldo José. Aspectos Epidemiológicos da Fibrose Cística. Revista do Hospital Universitário Pedro Ernesto, UERJ, Dezembro de 2011. Disponível em: http://bjhbs.hupe.uerj.br/WebRoot/pdf/70_pt.pdf. Acesso em: 02 de junho de 2021
3- KIM, C.A.; ALBANO, L.M.J.; BERTOLA, D.R. Genética na prática pediátrica 2a ed.. [Barueri, São Paulo]: Editora Manole, 2019. 9786555762419
4- RAMSEY, Bonnie W.; DAVIES, Jane; MCELVANEY, N. Gerard; TULLIS, Elizabeth; BELL, Scott C.; DřEVÍNEK, Pavel; GRIESE, Matthias; MCKONE, Edward F.; WAINWRIGHT, Claire E.; KONSTAN, Michael W.. A CFTR Potentiator in Patients with Cystic Fibrosis and theG551DMutation. New England Journal Of Medicine, [S.L.], v. 365, n. 18, p. 1663-1672, 3 nov. 2011. Massachusetts Medical Society
5- MIDDLETON, Peter G.; MALL, Marcus A.; DREVÍNEK, Pavel; LANDS, Larry C.; MCKONE, Edward F.; POLINENI, Deepika; RAMSEY, Bonnie W.; TAYLOR-COUSAR, Jennifer L.; TULLIS, Elizabeth VERMEULEN,François; MARIGOWDA, Gautham;MCKEE,Charlotte M. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. England: New England Journal of Medicine, November 7, 2019. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7282384/. Acesso em: 03 de junho de 2021.
6- HEIJERMAN, Harry G. M.; MCKONE, Edward F.; DOWNEY, Damian G.; BRAECKEL, Eva Van; ROWE, Steven M.; TULLIS, Elizabeth; MALL, Marcus A.; WELTER, John J.; RAMSEY, Bonnie W.; MCKEE, Charlotte M.; MARIGOWDA, Gautham; MOSKOWITZ, Samuel M.; WALTZ, David; SOSNAY, Patrick R.; SIMARD, Christopher; AHLUWALIA, Neil; XUAN, Fangjuan; ZHANG, Yaohua; TAYLOR-COUSAR, Jennifer L.; MCCOY, Karen S. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Volume 394. The Lancet Respiratory Medicine, November 23, 2019. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7571408/. Acesso em: 03 de junho de 2021.
7- RAMSEY, Bonnie W.; DAVIES, Jane; MCELVANEY, N. Gerard; TULLIS, Elizabeth. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. England: The New England Journal of Medicine, November 3, 2011. Disponível em: https://www.nejm.org/doi/full/10.1056/nejmoa1105185. Acesso em: 03 de junho de 2021.
8- TAYLOR-COUSAR, Jennifer L.; MUNCK, Anne; MCKONE, Edward F.; VAN DER ENT, Cornelis K.; MOELLER, Alexander; SIMARD, Christopher; WANG, Linda T.; INGENITO, Edward P.; MCKEE, Charlotte; LU, Yimeng; LEKSTROM-HIMES, Julie; ELBORN, J. Stuart. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. Volume 377. England: The New England Journal of Medicine, November 23, 2017. Disponível em: https://www.nejm.org/doi/pdf/10.1056/NEJMoa1709846. Acesso em: 4 de junho de 2021.
9- RATJEN, Felix; HUG, Christopher; MARIGOWDA, Gautham; TIAN, Simon; HUANG, Xiaohong; STANOJEVIC, Sanja; MILLA, Carlos E.; ROBINSON, Paul D.; WALTZ, David; DAVIES, Jane C. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Volume 5. The Lancet Respiratory Medicine, July 01, 2017. Disponível em: https://pubmed.ncbi.nlm.nih.gov/28606620/. Acesso em: 04 de junho de 2021.
10- WAINWRIGHT, Claire E.; ELBORN, J. Stuart; RAMSEY, Bonnie W.; MARIGOWDA, Gautham; HUANG, Xiaohong; CIPOLLI, Marco; COLOMBO, Carla; DAVIED, Jane C.; DE BOECK, Kris; FLUME, Patrick A.; KONSTAN, Michael W.; MCCOLLEY, Susanna A.; MCCOY, Karen; MCKONE, Edward F. ;MUNCK, Anne ;RATJEN, Felix; ROWE, Steven M.; WALTZ, David; BOYLE, Michael P. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. England: The New England Journal of Medicine, July 16, 2015. Disponível em: https://pubmed.ncbi.nlm.nih.gov/25981758/. Acesso em: 04 de junho de 2021.
11- DONALDOSON, Scott H.; PILEWSKI, Joseph M.; GRIESE, Matthias; COOKE, Jon; VISWANATHAN, Lakshmi; TULLIS, Elizabeth; DAVIES, Jane C.; LEKSTROM-HIMES, Julie A.; WANG, Linda T. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Volume 197. American Journal of Respiratory and Critical Care Medicine, January 15, 2018. Disponível em: https://pubmed.ncbi.nlm.nih.gov/28930490/. Acesso em: 05 de junho de 2021.
12- PEDRAZZI, Marco.; VERCELLONE, Silvia.; BARBERIS, Elettra.; CAPRARO, Michela.; DE TULLIO, Roberta.; CRESTA, Federico.; CASCIARO, Rosaria.; CASTELLANI, Carlo.; PATRONE, Mauro.; MARENGO, Emilio.; LECCA, Paola.; MELOTTI, Paola.; SORIO, Claudio.; MANFREDI, Marcello.; AVERNA, Monica. Identification of Potential Leukocyte Biomarkers Related to Drug Recovery of CFTR: Clinical Applications in Cystic Fibrosis. Volume 22. International Journal of Molecular Science, 10 April 2021. Disponível em: https://pubmed.ncbi.nlm.nih.gov/33920274/. Acesso em: 05 de junho de 2021.
13- SPOONHOWER, Kimberly A.; DAVIS, Pamela B.. Epidemiology of Cystic Fibrosis. Clinics In Chest Medicine, [S.L.], v. 37, n. 1, p. 1-8, mar. 2016. Elsevier BV. http://dx.doi.org/10.1016/j.ccm.2015.10.002. Acesso em: 06 de junho de 2021.
14- ROWE, Steven M.; MILLER, Stacey; SORSCHER, Eric J.. Cystic Fibrosis. New England Journal Of Medicine, [S.L.], v. 352, n. 19, p. 1992-2001, 12 maio 2005. Massachusetts Medical Society. Acesso em: 6 de junho de 2021.
¹Autor