REGISTRO DOI: 10.5281/zenodo.10725047
Aimê Armelin¹; Adonis Florença de Moraes¹; Diliane Primo¹; João Victor Tintori Gomes Mofato¹; Maria Emília Ventura Ziani Prestes¹; Mariana Leme Braga¹; Rafael Alvarenga de Oliveira Pereira¹; Gustavo Teixeira Grottone².
Palavras-chave: retinoblastoma; Infância; Incidência; Conscientização.
1. Introdução:
O retinoblastoma, embora seja câncer ocular primário mais comum em crianças, é relativamente raro em comparação com outros cânceres pediátricos, representando apenas entre 3 a 4% de todas as neoplasias malignas infantis. Sua incidência é de aproximadamente 1 em 14.000 a20.000 nascidos vivos, sem preferência por sexo. Aproximadamente dois terços é diagnosticado antes dos 2 anos, com 90% dos diagnósticos ocorrendo antes dos 5 anos.
Originado de células-tronco primitivas ou precursoras do cone na retina em desenvolvimento, o retinoblastoma está associado à inativação dos dois alelos do gene RB1, o primeiro gene supressor de tumor identificado. A pesquisa sobre esse câncer desempenhou um papel crucial nas descobertas em biologia do câncer, e avanços em genética molecular e terapia-alvo têm contribuído para melhorias significativas nas taxas de sobrevida infantil.
Nos países de alta renda (HICs), o retinoblastoma é considerado altamente curável, com uma taxa de sobrevida livre de doença próxima de 100%. No entanto, em países de baixa e média renda (PBMRs), onde mais de 80% dos casos ocorrem, o prognóstico é muitas vezes sombrio. A maioria dos casos de retinoblastoma é prevista para ocorrer na Ásia (53%), seguida pela África (29%), América Latina (8%), América do Norte (3%) e Europa (6%). Globalmente, a sobrevida para pacientes com retinoblastoma é estimada em 30%, refletindo as disparidades entre HICs e PBMRs.
O retinoblastoma, quando diagnosticado mais precocemente, tem capacidade de minimizar os prejuízos funcionais, cognitivos e psicológicos, principalmente para o paciente e também para os familiares.
2. Objetivos.
Avaliar os aspectos clínicos e as manifestações do retinoblastoma em crianças e abordar as formas de tratamento.
3. Metodologia
Foi realizada busca na base de dados (rodada em novembro de 2023) Medline (via PubMed);foram consultadas no Descritores de Ciência da Saúde (DeCS), além de, para a construção das linhas de pesquisa foram usadas como MeSh terms e seus sinônimos: “retinoblastoma”, “childhood” e “causes, treatments”. Após a busca, foi realizada a triagem e coleta de dados. Os títulos e resumos foram verificados e quanto aos critérios de inclusão foram selecionados somente estudos publicados em revistas médicas. Em seguida, os textos completos foram estudados, sendo selecionados ao todo 7 artigos.
4. Discussão
Retinoblastoma é um câncer de origem embrionário ou infantil que ocorre devido a perda conjunta dos pares do gene RB1, sendo este um supressor da formação do retinoblastoma. A célula de origem desse tumor é precursora dos fotorreceptores em cone, a qual perdeu ambos os alelos do gene supressor e permanece na camada nuclear interna da retina. Sendo assim, o câncer tem sua maior incidência em pacientes menores de 7anos visto que sua origem se deve a mutação de uma célula primitiva.
Em 50% dos pacientes diagnosticados pelo menos um par do alelo se encontra danificado em todas as células do organismo. Esse gene está presente no cromossomo 13q e codifica a proteína pRB (proteína retinoblastoma), que tem importante função na regulação do ciclo celular. A proteína bloqueia oE2F (promotor que suporta a divisão celular) impedindo com que a divisão aconteça de forma completa, sendo assim a falta dessa proteína desencadearia uma divisão incontrolada e possível mutação.
A causa primária dessa perda genética não pode ser definida com certeza, contudo diversos processos que danificam o DNA podem estar relacionados como raios X, raios ultravioleta, vírus e tabagismo. A melhor explicação até o momento refere uma predisposição familiar como causa mais provável, visto que ocorre em crianças e sua exposição a esses agentes externos é mínima. A linhagem dessa mutação pode dar início na espermatogênese, sendo a idade avançada do pai um fator importante que aumenta o risco de retinoblastomas.
Cerca de 98% dos pacientes tem ocorrência do retinoma (tumor benigno precursor) e posteriormente do retinoblastoma com a perda do gene RB1, entretanto, apenas 2% podem estar relacionados com mutações de outros genes, assim se tratando de possíveis outras causas para a incidência desse câncer, que por sua vez ainda é estatisticamente pequena.
Os primeiros sinais e sintomas do retinoblastoma são cruciais para um rápido diagnóstico e um tratamento prévio mais eficaz. Normalmente os primeiros sinais são reconhecidos pelos pais como um “brilho” diferente, caracterizado como leucopenia onde ocorre um reflexo anormal da pupila à luz, podendo ser identificado também oestrabismo3. Outros sinais e sintomas que podem ser identificados são: proptose que seria um deslocamento anterior do olho na cavidade da órbita ocular2, problemas na visão, aumento do tamanho dos olhos, dor nos olhos, vermelhidão na esclera e sangramentos na região anterior do olho.
Com o reconhecimento desses primeiros sinais e sintomas de um tumor ocular, cabe ao pediatra encaminhar acriança ao oftalmologista, que será responsável por fazer todos os testes, diagnóstico correto e o início do tratamento¹.
Dessa forma poderemos ter uma diminuição da porcentagem de crianças que perdem a visão devido um diagnóstico tardio e melhorar a qualidade de vida dessas crianças.
O início do diagnóstico do retinoblastoma pode ser feito por um médico generalista através da inspeção ocular de propedêutica simples: o sinal comum é a leucocoria (“pupila branca”) popularmente conhecido como “olho de gato” e quando relatado pelos responsáveis reflexo ocular atípico nos olhos da criança, deve-se colocar o retinoblastoma como principal hipótese e encaminhar o paciente para um oftalmologista afim de definir a patologia presente. Em casos mais avançados, pode-se observar no paciente o estrabismo com acometimento da visão central , alteração na coloração da íris e aumento do olho e da córnea decorrentes do aumento da pressão e inflamação ocular não infecciosa³ “descolamento de retina com líquido sub-retiniano associado, semeadura sub-retiniana ou sementes dentro do gel vítreo³.
Para diagnosticar a doença, o oftalmologista deve se atentar em qual método diagnóstico utilizará. A Biópsia é contra-indicada a ser feita, pois apresenta risco de disseminação tumoral assim como tomografias computadorizadas e “varreduras são evitadas porque a radiação pode induzir disseminação em cânceres primários em pessoas portadoras de mutações RB 1105″3. Dessa forma, a oftalmoscopia indireta com a pupila farmacologicamente dilatada é geralmente suficiente para a detecção. A ultrassonografia ocular (b-scan) é utilizada para verificar calcificação, algo típico no retinoblastoma. A ressonância magnética pode ser utilizada para estadiamento⁴ e para avaliar a invasão da óptica do nervo 74 ocular e a presença de retinoblastoma trilateral (pinealoblastoma e tumores intracranianos neuroectodérmicos primitivos associados a mutações do gene RB1)74,75.
Porém, para melhor reconhecimento da doença é necessário um exame detalhado da retina sob anestesia geral para distinguir os diagnósticos diferenciais (doença de Coats, vasculatura fetal persistente e hemorragia vítrea) e classificar a gravidade da doença intraocular. O desenho preciso do fundo de olho é essencial para mapear a carga e a localização do tumor ;e, se possível, a utilização da grande angular, câmera de fundo de olho portátil utilizada para visualizar e gravar toda a retina³. Alta frequência (50 MHz), biomicroscopia ultrassônica 77 eOCT 63 são úteis para descobrir tumores invisíveis em bebês com doença familiar³. A boa imagem e documentação de toda a retina apoia a classificação ocular do estadiamento, auxilia na análise de progressão e regressão tumoral, além de evidenciar as melhores opções de tratamento em cada caso³.
O tratamento do retinoblastoma evoluiu durante os anos com investimento em novas tecnologias e pesquisas, a escolha do mesmo varia conforme o estadiamento e a presença de manifestações extra oculares e deve ser realizado com uma equipe multidisciplinar⁵.
A terapêutica tem como principais objetivos a preservação da visão assim como a prevenção de doenças metastáticas. Dentre as principais opções estão: enucleação, radioterapia, quimioterapia sistêmica eintra-arterial, braquiterapia, laserterapia e crioterapia.
Algoritmo de tratamento para retinoblastoma baseado na lateralidade e no estágio da Classificação Internacional de Retinoblastoma (ICRB):
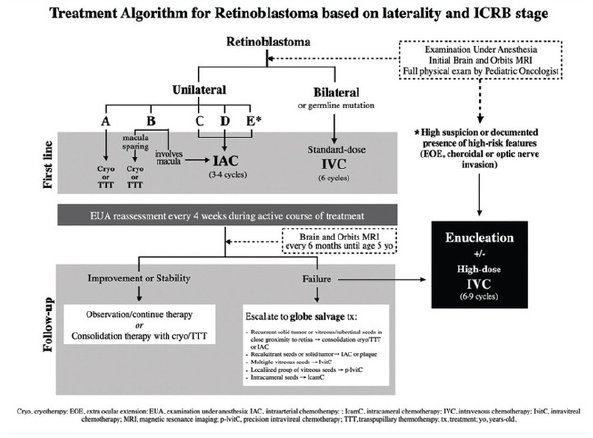
Apesar das modernas terapias a enucleação ainda é comum, indicada principalmente nos tumores de grau E(tumor extensivo) e quando há má visualização das margens da lesão, consiste basicamente na retirada do globo ocular e tem como principais complicações granuloma piogênico, cistos conjuntivais eblefaroptose.
A braquiterapia é indicada nos casos de tumores médios e nos casos de recorrência após quimioterapia intravenosa (IVC) ou quimioterapia inter-arterial (IAC). Na braquiterapia coloca-se o material radioativo em uma placa de ouro ou de chumbo para proteger os tecidos adjacentes, depois de 2a4dias retira-se a mesma em centro cirúrgico. Dentre os principais efeitos colaterais estão catarata e maculopatia por radiação.
A quimioterapia intravenosa (IVC) tem como principal pilar a redução do volume tumoral assim permitindo optar por terapias conservadoras como a crioterapia. As indicações são nos casos de retinoblastoma bilateral e casos de acometimento do nervo óptico. Os quimioterápicos mais usados na IVC são vincristina, vepeside e carboplatina (VEC) os mesmos são administrados através de um microcatéter na artéria oftálmica. Há relatos em que administração da crioterapia antes da quimioterapia aumenta a disponibilidade do medicamento nos espaços intra-oculares quando administrada nas primeiras 48h pós resfriamento⁵.
Na quimioterapia intra-arterial (IAC) existe a possibilidade da administração seletiva, uma dose única na maioria das vezes, e é maior quando comparada a IVC, porém essa linha de tratamento apresentou dificuldades quanto ao suprimento da artéria oftálmica acometida pelo retinoblastoma. Porém se concluiu ser uma terapia segura e efetiva nos casos de retinoblastoma intra-ocular avançado⁶.
Por fim, a crioterapia é indicada no tratamento de pequenos tumores e costuma ser associada com as quimioterapias, sendo a principal delas a IVC atuando de forma combinada para reduzir as células tumorais. Ela é realizada através da oftalmoscopia indireta, com auxílio da sonda de crioterapia acontece o congelamento das lesões. A principal complicação da crioterapia é o descolamento de retina⁵.
Em suma o tratamento do retinoblastoma depende da idade, do grau de estadiamento do tumor e da disponibilidade de recursos terapêuticos, assim como deve ser levado em consideração o prognóstico e a preservação da acuidade visual. Portanto, sua escolha deve ser individualizada de acordo com a realidade de cada paciente⁶.
5. Conclusão
O retinoblastoma é um câncer ocular que afeta principalmente crianças menores de 7anos, e que se origina da perda de um gene supressor de tumores chamado RB1. Esse gene é responsável por controlar o ciclo celular e impedir a divisão desordenada das células precursoras dos fotorreceptores da retina. A causa mais comum dessa perda é uma predisposição familiar, mas outros fatores ambientais podem estar envolvidos. O diagnóstico precoce é essencial para o tratamento eficaz e a preservação da visão. Os principais sinais e sintomas são a leucocoria, o estrabismo, a alteração da cor da íris, o aumento do olho e a dor ocular. O médico oftalmologista é o profissional indicado para realizar os exames e confirmar o diagnóstico, bem como iniciar o tratamento adequado.
Para diagnosticar o retinoblastoma, o oftalmologista deve realizar um exame oftalmológico detalhado, que pode incluir oftalmoscopia indireta, ultrassonografia, ressonância magnética e exame sob anestesia geral. O objetivo é identificar o tumor, descartar outras doenças e classificar a gravidade da doença. O tratamento depende da lateralidade, do estágio e da presença de metástases. As opções terapêuticas incluem enucleação, radioterapia, quimioterapia, braquiterapia, laserterapia e crioterapia. O principal objetivo é preservar a visão e prevenir a disseminação do câncer.
O texto aborda as diferentes modalidades de tratamento do retinoblastoma, explicando as indicações, as vantagens e as desvantagens de cada opção terapêutica. O texto destaca que o tratamento deve ser individualizado de acordo com a idade, o estágio e o prognóstico do tumor, bem como a preservação da visão. O texto também ressalta a importância de uma equipe multidisciplinar para o acompanhamento dos pacientes. O retinoblastoma é uma doença grave, mas que pode ser tratada com sucesso se diagnosticada e tratada adequadamente.
6. Referências
1.Antoneli CBG, Steinhorst F, Ribeiro Kde CB, Chojniak MMM, Novaes PER, Arias V, et al.. Opapel do pediatra no diagnóstico precoce do retinoblastoma. Rev Assoc Med Bras [Internet]. 2004 Oct;50(4):400–2. Disponível em: https://doi.org/10.1590/S0104-42302004000400030
2.Moin M, Malik TG, Siddiq L. Clinical Patterns And Outcomes Of Retinoblastoma In ATertiary Care Centre Of ADeveloping Country. JPak Med Assoc. 2023 Sep;73(9):1881-1883. doi: 10.47391/JPMA.7689. PMID: 37817703.
3.Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Meunier FL, et al. Retinoblastoma. Nature Reviews Disease Primers [Internet]. 2015 Aug 27;1(1). Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5744255/
4.Jenkinson H. Retinoblastoma: diagnosis and management—the UK perspective. Arch Dis Child [Internet]. 4maio 2015 [citado 12 nov 2023];100(11):1070-5. Disponível em: https://doi.org/10.1136/archdischild-2014-306208
5. Ancona-Lezama, David; Dalvin, Lauren A1 ; Escudos, Carol L2 , . Tratamento moderno do retinoblastoma: Uma revisão de 2020. Indian Journal of Ophthalmology 68(11):p 2356-2365, novembro de 2020. | DOI: 10.4103/ijo.IJO_721_20
6. Revista Educação em saúde. Análise das diferentes abordagens da quimioterapia no tratamento de retinoblastoma- RESU –Revista Educação em Saúde: V7, suplemento 1,2019
7. Soliman SE, Racher H, Zhang C, MacDonald H, Gallie BL. Genetics and Molecular Diagnostics in Retinoblastoma–An Update. Asia Pac JOphthalmol (Phila). 2017 Mar-Apr;6(2):197-207. doi: 10.22608/APO.201711. PMID: 28399338.
¹Acadêmico de Medicina, Universidade Metropolitana de Santos (UNIMES).
²Preceptor da liga acadêmica de oftalmologia da Universidade Metropolitana de Santos (UNIMES).