PROGNOSIS OF POMPE DISEASE IN PATIENTS UNDERGOING ENZYME REPLACEMENT THERAPY: A SYSTEMATIC REVIEW
REGISTRO DOI: 10.5281/zenodo.10028042
Lorena de Souza Bahia¹
Pedro Vinícius Costa Barreto²
Ana Jovina Barreto Bispo³
RESUMO
INTRODUÇÃO: A doença de Pompe é uma condição genética metabólica que cursa com comprometimento neuromuscular. Seu caráter progressivo requer um reconhecimento e tratamento precoces, almejando um melhor desfecho aos seus portadores.
OBJETIVO: Avaliar a influência da introdução precoce da terapia de reposição enzimática nas funções cardíaca e respiratória, bem como no desenvolvimento neuropsicomotor e expectativa de vida útil.
FONTE DOS DADOS: Trata-se de uma revisão sistemática que buscou artigos não pagos indexados na PubMed®, Scielo, MedLine e Scopus nos últimos cinco anos, utilizando os seguintes descritores em inglês e seus correspondentes em português “Prognosis“, “Storage“, “Disease“, “Glycogen” e “Type II“, correlacionados por intermédio do operador booleano AND.
SÍNTESE DOS DADOS: Um total de vinte e nove artigos foi encontrado, dos quais apenas quatro foram incluídos nesse estudo. Vinte e quatro foram excluídos após leitura do resumo, por não se enquadrarem aos critérios de elegibilidade. Todos os artigos selecionados demonstraram o efeito da introdução precoce da terapia enzimática em desacelerar a progressão da doença, além de possibilitar a reversão da hipertrofia ventricular. Os efeitos sobre a função respiratória foram inconclusivos, existindo duas vertentes: uma que aborda a manutenção da função respiratória e outra que destaca que, independente do tratamento, alguns pacientes apresentarão declínio em parâmetros de avaliação pulmonar, como capacidade vital forçada.
CONCLUSÕES: Está claro o impacto do atraso na terapia enzimática no comprometimento do prognóstico dos pacientes, o que aumenta a responsabilidade dos profissionais em efetuarem o diagnóstico e o tratamento precoces, possibilitando uma melhor qualidade de vida às crianças.
Palavras-chave: Prognóstico; Doença de depósito de glicogênio tipo II; Terapia de reposição de enzimas.
ABSTRACT
INTRODUCTION: Pompe disease is a genetic metabolic condition that leads to neuromuscular impairment. Its progressive nature requires early recognition and treatment, aiming for a better outcome for its sufferers.
OBJECTIVE: To evaluate the influence of early introduction of enzyme replacement therapy on cardiac and respiratory functions, as well as neuropsychomotor development and life expectancy.
DATA SOURCE: This is a systematic review that searched for unpaid articles indexed in PubMed®, Scielo, MedLine and Scopus in the last five years, using the following descriptors in English and their corresponding Portuguese “Prognosis”, “Storage”, “Disease”, “Glycogen” and “Type II”, correlated using the Boolean operator AND.
DATA SYNTHESIS: A total of twenty-nine articles were found, of which only four were included in this study. Twenty-four were excluded after reading the abstract, as they did not meet the eligibility criteria. All selected articles demonstrated the effect of early introduction of enzyme therapy in slowing the progression of the disease, in addition to enabling the reversal of ventricular hypertrophy. The effects on respiratory function were inconclusive, with two aspects: one that addresses the maintenance of respiratory function and another that highlights that, regardless of the treatment, some patients present impairment in lung assessment parameters, such as forced vital capacity.
CONCLUSIONS: The impact of the delay in enzymatic therapy on the worsening of the patients’ prognosis is clear, which increases the responsibility of professionals to carry out an early diagnosis and treatment, guaranteeing a better quality of life for children.
Keywords: Prognosis; Glycogen storage disease Type II; Enzyme Replacement Therapy
INTRODUÇÃO
Intitulada como doença do armazenamento do glicogênio do tipo II, a doença de Pompe é uma rara doença genética metabólica, autossômica recessiva, que cursa com progressivo comprometimento neuromuscular.1 É causada pela deficiência parcial ou total da enzima maltase-ácida (ou alfa 1-4 glicosidase ácida), decorrente de uma mutação no gene que a codifica, o qual localiza-se no cromossomo 17q25.2.1 Essa enzima é responsável pela degradação de glicogênio em glicose, processo esse comprometido na doença de Pompe (DP), o que leva ao acúmulo de glicogênio nas células miocárdicas, nas musculaturas lisa e esquelética e no sistema nervoso central.1,2 Dados referentes à epidemiologia dessa doença ainda são incertos, havendo variações a depender da população estudada.3 Contudo, estima-se uma incidência da doença em torno de 1:40.000 norte-americanos.4 Muitas são as manifestações clínicas da doença. Didaticamente ela é classificada em: DP de início infantil clássica e não clássica e DP de início tardio (Quadro 1).1,2
Quadro 1- Formas de Apresentação Clínica da Doença de Pompe
CLASSIFICAÇÃO DP de início infantil: forma clássica DP de início tardio: forma não clássica INÍCIO DOS SINTOMAS Primeiro ano de vida A partir do segundo ano de vida SINTOMAS Fraqueza muscular generalizada grave, hipotonia, cardiomiopatia hipertrófica, evoluindo para insuficiência respiratória Inicialmente cursa com fraqueza muscular em cintura pélvica, com posterior acometimento da musculatura axial e diafragmática, podendo levar a insuficiência respiratória; não há envolvimento cardíaco ATIVIDADE ENZIMÁTICA DA GAA <1% 1 – 30% PROGRESSÃO DA DOENÇA Progressão mais rápida da doença Progressão mais lenta da doença
Em geral, a forma clássica da DP de início infantil, manifestada ainda no primeiro ano de vida, apresenta maior gravidade e pior prognóstico, principalmente se o tratamento não for introduzido precocemente.1 A atividade enzimática quase nula (<1%) da alfa-1,4-glicosidase ácida (GAA), está diretamente relacionada a esse prognóstico desfavorável, cursando com quadros graves de fraqueza muscular, hipotonia e cardiomiopatia, podendo evoluir para insuficiência respiratória e óbito nos primeiros dias a semanas de vida.2 Outros sintomas menos característicos podem estar presentes, como a macroglossia e hepatomegalia.1,2 A principal diferença para a DP de início infantil não clássica está na ausência de envolvimento cardíaco.5
Por último, mas não menos importante, a DP de início tardio, pode manifestar-se ainda no primeiro ano de vida ou mais tardiamente, na sexta década de vida.1 Observa-se uma progressão mais lenta dos sintomas e um quadro inicial bastante heterogêneo, variando de fraqueza muscular em cintura pélvica a um predomínio de fraqueza da musculatura respiratória e axial.1 Mesmo que, inicialmente o paciente não apresente comprometimento respiratório, o progredir da doença inerente levará à insuficiência respiratória e óbito na maioria dos casos.1
O diagnóstico tardio de doenças raras, em especial daquelas que com progressivo comprometimento neuromuscular, é responsável por retardar o tratamento. Diante dessa situação, métodos de triagem têm sido desenvolvidos, em prol da identificação de pacientes com suspeita de DP, prosseguindo com a investigação diagnóstica caso a suspeita persista. Esse processo tem início com a triagem dos casos suspeitos, por meio da coleta de amostra de sangue em papel-filtro e confirmação do diagnóstico com a mensuração do percentual de GAA nos leucócitos e/ou fibroblastos e genotipagem da enzima GAA, em busca de mutações patogênicas.1,2
Desde 2006, com a aprovação, pela Food and Drug Administration (FDA), da terapia de reposição enzimática (TRE) com o uso da rhGAA, os pacientes diagnosticados com a DP tiveram seu prognóstico modificadoe a esperança de vida ressurgiu.6 Apesar dos notórios benefícios da aplicação da TRE, esta não é capaz de reverter a doença já instalada.6,7,8 Na imensa maioria dos casos, esses indivíduos serão submetidos ao tratamento por anos, sendo posteriormente conduzidos aos cuidados paliativos.2 Reitera-se, portanto, a importância da inclusão da família na tomada de decisões e da presença de uma equipe multidisciplinar, que conte com médicos, fisioterapeutas, fonoaudiólogos, terapeutas ocupacionais, psicólogos, dentre outros profissionais.
Ainda com relação à TRE, sabe-se da influência de diversos fatores na individualizada resposta ao tratamento, tais como: idade de início da terapia de reposição enzimática, severidade da cardiomiopatia, tratamento prévio devido a patologia muscular e o status MIRC (material imunológico medido pela reação cruzada), também conhecido pela sua sigla em inglês CRIM (cross-reaction immunological material).2,6,9 No que tange ao CRIM, definido com base em um estudo genético que identifica o número de variantes associadas à doença, sabe-se que é um importante preditor do prognóstico na DP de início infantil.8,10 Pacientes CRIM negativo caracterizam-se por portarem duas variantes da GAA, o que leva à ausência de qualquer atividade enzimática residual.2 Não obstante, caracterizam-se por formular respostas imunológicas que vão de encontro à ação da TRE, o que leva a um pior prognóstico.2,10 No caso dos pacientes CRIM positivo, observa-se melhor desfecho clínico e uma melhor e mais promissora resposta à TRE, afinal, esses pacientes apresentam algum grau de atividade enzimática residual e, sua resposta imunológica é branda.11
Esta revisão sistemática objetivou avaliar a influência da introdução precoce da terapia de reposição enzimática (TRE) nas funções cardíaca e respiratória de crianças e adolescentes portadores da doença de Pompe, bem como no seu desenvolvimento neuropsicomotor e expectativa de vida útil.
METODOLOGIA
Este é um estudo de revisão sistemática, conduzido de acordo com as diretrizes Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA).
Foram incluídos estudos não pagos observacionais comparativos, revisões sistemáticas e relatos de casos nos idiomas inglês, português e espanhol, publicados no período de 2018 a 2023, que tratem sobre o efeito da introdução precoce da terapia de reposição enzimática no prognóstico dos pacientes acometidos pela Doença de Pompe, na faixa etária de 0 a 18 anos. Para sua construção, foi feita a busca de artigos indexados na PubMed®, Scielo, MedLine e Embase, utilizando os seguintes descritores em inglês (MeSH) e seus correspondentes em português (DeCS) “Prognosis”, “Storage”, “Disease”, “Glycogen” e “Type II”, correlacionados por intermédio do operador booleano AND. A busca ativa de artigos foi realizada entre os meses de abril, maio e junho de 2023.
Foram excluídas metanálises e estudos multicêntricos. Além disso, também foram excluídos artigos que abordavam o efeito da introdução da TRE nos pacientes com início dos sintomas e/ou diagnóstico da DP na idade adulta ou mesmo em portadores de formas atípicas da doença (sem comprometimento da musculatura esquelética, por exemplo).
Para o processo de seleção de estudos, houve a participação de dois revisores, os quais efetuaram o processo de triagem e seleção dos estudos de maneira independente e individualizada. In the first stage, the articles found were read and selected based on their titles and abstracts, as well as on their adequacy to the previously established inclusion and exclusion criteria. In the second stage, the selected articles were read in their entirety, and their suitability to the study objectives was reevaluated based on the article’s main objectives, study design, and sample characteristics.
RESULTADOS
Inicialmente, encontrou-se 29 artigos indexados em bases de dados, dos quais 15 foram encontrados na PubMed®, 5 na MedLine, 7 na Embase e 2 na Scielo. Após aplicação dos critérios de elegibilidade, 23 artigos foram excluídos e 6 foram selecionados para leitura do título. Isso resultou em 6 artigos selecionados para leitura completa do texto. Desses, dois foram excluídos, um pela indisponibilidade do artigo completo e outro por não corresponder ao objetivo dessa revisão, ao abordar o efeito do treinamento muscular respiratório nos pacientes portadores da doença em sua forma infantil. Por conseguinte, 4 artigos foram oficialmente selecionados para o processo de construção da revisão sistemática. O processo de seleção dos artigos com base nos critérios de elegibilidade está descrito no PRISMA flow diagram (Figura 1).
Figura 1– Fluxograma do processo de inclusão dos estudos, segundo PRISMA 2020
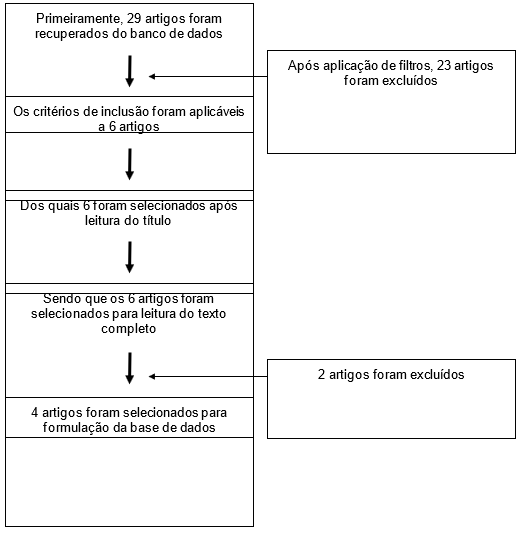
Não foram encontrados artigos publicados nos anos de 2018, 2021 e 2023. Considerando-se os artigos selecionados, 25% da amostra foi publicada em dezembro de 2019, 50% em 2020 (nos meses de março e setembro) e 25% foram publicados em dezembro de 2022. Dentre os artigos selecionados, 2 eram relatos de casos, 1 consistia em uma revisão retrospectiva de prontuários e, por fim, 1 era um ensaio clínico randomizado (Quadro 2).
Quadro 2 – Tipo de estudos e tamanho da amostra
Autor Tamanho da Amostra (n) Tipo de Estudo GUPTA P, et al. 1 Relato de caso ELMALLAH MK, et al. 14 Revisão retrospectiva de prontuário BOLANO-DIAZ C; DIAZ-MANERA J – Ensaio clínico randomizado TOLANI D., et al. 2 Relato de caso
Fonte: autoria própria
Os desfechos estudados em cada artigo selecionado encontram-se no quadro 3.
Quadro 3 – Desfechos estudados nos estudosselecionados
Artigos selecionados Retardo na progressão da doença Impacto na sobrevida Aquisição de marcos do desenvolvimento Melhoria da hipertrofia cardíaca Impacto na função respiratória Gupta P, et al. X X Elmallah MK, et al. X X Bolano-Diaz C, Diaz-Manera J X X X X X Tolani D, et al. X X
Fonte: autoria própria
O artigo redigido por Tolani D et al. não cita a idade ao diagnóstico, apenas que esse foi feito após o nascimento. O artigo escrito por Elmallah MK et al., por sua vez não apresenta citações sobre ecocardiograma e/ou eletrocardiograma. A descrição metodológica encontra-se no quadro 4.
Quadro 4 – Descrição metodológica dos estudos incluídos na revisão sistemática
Artigo Idade média ao diagnóstico Status CRIM1 Ecocardio- grama Eletrocardio-grama Tratamento Prognóstico GUPTA, P. Pré natal Negativo Antes do nascimento (31 semanas de idade gestacional): discreta hipertrofia ventricular esquerda afetando septo interven- tricular Após nascimento (1º dia de vida): severa hipertrofia biventricular Após o nascimento: ritmo sinusal; hipertrofia biventricular ITI2 – 2º dia de vida TRE3 – 3º dia de vida (dose: 20 mg/kg; infusões quinzenais) Completa recuperação da função cardíaca na 11ª semana de vida; Velocidade normal de crescimento; Percentil 90 para peso e estatura aos 4 anos de idade; Desenvolvimento adequado para a idade ELMALLAH, M.K. 2 meses Positivo Não foi informado Não foi informado Média de idade de introdução aos 3 meses; Doses foram variáveis para cada paciente Declínio da capacidade vital forçada nas posições ereta e supina TOLANI, D. Após nascimento Positivo Após nascimento: hipertrofia biventricular com normalidade anatômica Antes do nascimento: significativa hipertrofia biventricular Não efetuado 5 meses: infecção de via aérea superior; intubação orotraqueal; ventilação mecânica Evoluiu necessitando de traqueostomia 20 meses: Parada cardiorrespiratória com insuficiência respiratória hipóxica aguda Após nascimento Positivo Após nascimento: moderada hipertrofia biventricular com função biventricular levemente deprimida e normalidade anatômica Antes do nascimento: arritmias fetais TRE – 11º dia de vida (dose: não especificada; infusões quinzenais) 17 meses: função cardíaca normal, com completa resolução da hipertrofia biventricular BOLANO-DIAZ C. e DIAZ-MANERA J. A TRE teve um impacto considerável na progressão da doença, especialmente no caso da IOPD4
2ITI – indutor de tolerância imunológica
3TRE – terapia de reposição enzimática
4IOPD – infantile onset Pompe disease
DISCUSSÃO
Gupta P et al. destacou o impacto do tratamento na sobrevida dos pacientes, ao enfatizar que, previamente à sua implantação, a morte era iminente nos primeiros dois anos de vida, na maioria das vezes em virtude de falência cardiorrespiratória. Apesar de ter modificado o curso da doença, inclusive com melhora da sobrevida, o autor em questão reitera que a TRE é incapaz de reverter patologias de base e está longe de ser a cura para a condição, tendo-se em vista que um considerável número de pacientes continua evoluindo ao longo dos anos, apesar do tratamento.12
Tolani D et al. e Gupta P et al. destacaram a relação da introdução precoce da TRE com a recuperação da função cardíaca. Tolani D et al. descreveu em seu estudo, dois casos clínicos, ambos CRIM positivos, com detecção de anormalidades cardíacas ainda no pré-natal, cujo prognóstico foi completamente distinto, decorrente do atraso para início da terapia. Aquele que teve a TRE iniciada ao 11º dia de vida, apresentou completa resolução do quadro de hipertrofia ventricular aos 17 meses. Não obstante, o outro paciente obteve prognóstico desfavorável. O artigo não especifica a idade de início, porém, o relato dos fatos nos leva à compreensão de que ocorreu em uma fase em que já havia comprometimento de órgãos e que o diagnóstico tardio levou ao postergar do início da TRE. O referido paciente evoluiu com quadro de infecção de vias aéreas superiores, necessitando de ventilação invasiva, além de traqueostomia. Por fim, deu entrada a hospital em franco quadro de parada cardiorrespiratória, evoluindo para óbito.13 Gupta P et al., por sua vez, ilustrou o primeiro paciente diagnosticado ainda no pré-natal como portador da DP, CRIM negativo, submetido a profilaxia com indutor de tolerância imunológica com Rituximabe ao 2º dia de vida, além de TRE no 3º dia de vida. Embora isso abra um leque de discussões sobre a influência do status CRIM na responsividade dos pacientes ao tratamento, que não é o objetivo desse estudo, não se pode negar que esse paciente obteve bons resultados devido ao início precoce da terapia.12 Em se tratando da função cardíaca, observou-se completa recuperação, além de resolução da hipertrofia ventricular por volta da 11ª semana de vida. O efeito também foi demonstrado a longo prazo e o paciente não apresentou novas alterações em exames cardiológicos, realizados periodicamente a cada 3 meses.12
Com relação ao efeito da administração precoce da TRE na função respiratória, ElMallah MK et al. dedicaram esforços na realização de uma revisão retrospectiva de prontuários que contou com 14 pacientes portadores da DP de início infantil, todos CRIM positivos. Chegaram à conclusão de que a TRE foi capaz de manter a função respiratória de alguns pacientes, enquanto outros continuaram apresentando declínio ao longo dos anos. Dentre os parâmetros avaliados, tem-se a Capacidade Vital Forçada (CVF), a qual apresentou declínio em todos os pacientes, quando esses estavam em posição ereta e supina. Outrossim, observou-se que todos os pacientes com melhorias na CVF tiveram a TRE iniciada nos primeiros 3 meses de vida, o que reafirma o impacto do tratamento precoce na funcionalidade respiratória. Estes autores relataram também que em crianças com diagnóstico e tratamento tardios, a CVF do paciente permaneceu estável, apesar de baixa. Ele sugeriu ainda que a ineficiência da TRE no neurônio motor respiratório reside no direcionamento e absorção ineficaz da enzima através da musculatura respiratória. Importante destacar que os casos descritos por ElMallah MK et al. foram de indivíduos com média de idade ao diagnóstico de 2 meses e de início da terapia aos 3 meses, com uma variação de 18 horas a 42 meses de vida.14
Em sua revisão, Bolano-Diaz C e Diaz-Manera J destacaram que, desde meados de 2006, com o alavancar dos estudos acerca do tratamento da DP, não há evidências que comprovem seu efeito sobre a funcionalidade respiratória e esquelética. Os estudos incluídos na construção de seu ensaio clínico demonstraram tanto pacientes com significativa melhora, como aqueles com alcance de limitados benefícios.15 Descreveu ainda um estudo observacional realizado na Europa, que avaliou o impacto de diferentes regimes de TRE (baixa e alta dosagem) na sobrevida e no desenvolvimento de 116 portadores da DP de início infantil. Com relação ao impacto na sobrevida, a conclusão foi de que pacientes que são submetidos a regimes de altas dosagens (infusões semanais de 40 mg/kg) têm uma melhor taxa de sobrevida em 5 anos, informação essa não observada entre aqueles submetidos a baixas dosagens (infusões semanais de 20 mg/kg). No que tange ao efeito sobre marcos motores, observou-se que 53% dos pacientes que receberam regime de baixa dose e 93% dos que receberam altas doses, foram capazes de aprender a andar. Todavia, ainda não há evidências concretas sobre o efeito da TRE nesse parâmetro, sendo necessários novos estudos que abordem esse tópico.15
Pode-se reconhecer o benefício da TRE no desfecho clínico dos pacientes diagnosticados com DP de início infantil. O início precoce da terapia foi essencial em retardar a progressão da doença e em promover melhorias no quadro sintomatológico. Evidências demonstram que um atraso esteve associado a piores resultados biológicos, físicos e no desenvolvimento. Contudo, é importante destacar que se observou que o efeito da TRE se torna imprevisível caso a introdução seja feita após o 5º mês de vida.12,13
CONSIDERAÇÕES FINAIS
Conclui-se desse modo que novos estudos de maior qualidade precisam ser desenvolvidos, com o objetivo de avaliar o impacto da introdução precoce da TRE na função respiratória e na aquisição de marcos motores.
REFERÊNCIAS
- Kleigman RM, 3rd St Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM. Nelson Tratado de Pediatría. 21ed.Barcelona: Elsevier; 2020.
- Junior JCL, Nascimento OJM, Oliveira ASB, Junior METD, Marrone CD, Siqueira HH, et al. Guidelines for the diagnosis, treatment and clinical monitoring of patients with juvenile and adult Pompe disease. Arq Neuropsiquiatr. 2016;74(2):166-176.
- Palahuta HV, Fartushna OY, Selina OG, Fartushnyi YM, Koval TV. Glycogen storage disease type II a narrative literature review and a case report of late-onset Pompe disease in a young white child. Wiad Lek. 2021;74(4):1032-1036.
- Morales A, Anilkumar AC. Glycogen Storage Disease Type II. StatPearls. Treasure Island (FL); 2023.
- Lim JA, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci. 2014;6:177.
- Do HV, Khanna R, Gotschall R. Challenges in treating Pompe disease: an industry perspective. Ann Transl Med. 2019 Jul; 7(13):291.
- Byrne BJ, Fuller DD, Smith BK, Clement N, Coleman K, Cleaver B, et al. Pompe disease gene therapy: neural manifestations require consideration of CNS directed therapy. Ann Transl Med. 2019; 7(13): 290.
- Abbott MA, Prater SN, Banugaria SG, Richards SM, Young SP, Rosenberg AS, et al. Atypical immunologic response in a patient with CRIM-negative Pompe disease. Mol Genet Metab. 2011;104(4): 583-586.
- Crisp KD, Case LE, Kravitz RM, Kishnani PS, Jones HN. Training, detraining, and retraining: Two 12-week respiratory muscle training regimens in a child with infantile-onset Pompe disease. J Pediatr Rehabil Med. 2020; 13(1): 71-80.
- Poelman E, Hoogeveen-Westerveld M, Kroos-de Haan MA, Van den Hout JMP, Bronsema KJ, Van de Merbel NC, et al. High Sustained Antibody Titers in Patients with Classic Infantile Pompe Disease Following Immunomodulation at Start of Enzyme Replacement Therapy. J Pediatr. 2018;195:236-243.e3.
- Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99(1):26-33.
- Gupta P, Shayota BJ, Desai AK, Kiblawi F, Myridakis D, Messina J, et al. A Race Against Time—Changing the Natural History of CRIM Negative Infantile Pompe Disease. Frontiers in Immunology. 2020; 1929.
- Tolani D, Bansal N, Sehgal S. Infantile-onset pompe disease: a tale of two cases. Cardiol Young. 2020; 30(2):275-277.
- ElMallah MK, Desai AK, Nading EB, DeArmey S, Kravitz RM, Kishnani PS. Pulmonary Outcome Measures in Long Term Survivors of Infantile Pompe Disease on Enzyme Replacement Therapy: A case series. Pediatr Pulmonol. 2020;55(3):674–681.
- Bolano-Diaz C, Diaz-Manera J. Therapeutic Options for the Management of Pompe Disease: Current Challenges and Clinical Evidence in Therapeutics and Clinical Risk Management. Ther Clin Risk Manag. 2022;18:1099-1115.
- Cohen JL, Chakraborty P, Fung-Kee-Fung K, Schwab ME, Bali D, Young SP, et al. In Utero Enzyme-Replacement Therapy for Infantile-Onset Pompe’s Disease. N Engl J Med. 2022; 387(23): 2150-2158.
- Pitts T, Bordelon R, Huff A, Byrne BJ, Smith BK. Cough Effectiveness and Pulmonary Hygiene Practices in Patients with Pompe Disease. Lung. 2019;197(1):1-8.
- Owens P, Wong M, Bhattacharya K, Ellaway C. Infantile-onset Pompe disease: A case series highlighting early clinical features, spectrum of disease severity and treatment response. J Paediatr Child Health. 2018;54(11):1255-1261.
- Ullah A, Zubaida B, Cheema HA, Naeem M. Identification of two novel variants in GAA underlying infantile-onset Pompe disease in two Pakistani families. J Pediatr Endocrinol Metab. 2020;33(4):553-556.
- Codazzi AC, Ippolito R, Novara C, Tondina E, Cerbo RM, Tzialla C. Hypertrophic cardiomyopathy in infant newborns of diabetic mother: a heterogeneous condition, the importance of anamnesis, physical examination and follow-up. Ital J Pediatr. 2021;47(1):197.
- Zhang Y, Zhang C, Shu JB, Zhang F. Atypical infantile-onset Pompe disease with good prognosis from mainland China: A case report. World J Clin Cases. 2022 Apr 6;10(10):3278-3283.
¹ORCID: 0009-0009-0835-7673
Universidade Tiradentes
Aracaju-Sergipe-Brasil
²ORCID: 0000-0003-1546-4708
³ORCID: 0000-0002-6228-768X
Universidade Tiradentes
Professora Assistente
Aracaju-Sergipe-Brasil