REGISTRO DOI: 10.5281/zenodo.7812336
Raul Dias Kezan
RESUMO
Descoberta em 1996, a mutação do gene CCR5 se mostrou capaz de conferir resistência à infecção pelo HIV da população. Isso levou a uma intensa pesquisa na tentativa de utilizar essa descoberta como base para uma terapia curativa para o HIV, tendo em vista que a maioria das células infectadas pelo vírus HIV expressam o receptor CCR5 em sua superfície. Nesse sentido, este artigo científico de revisão sistemática busca analisar os estudos disponíveis que investigaram o uso de terapias baseadas na destruição ou bloqueio do receptor CCR5 como uma possível cura para o HIV. A descoberta do papel do gene CCR5 e do efeito protetor da mutação despertam um crescente interesse pelo papel terapêutico que o possível bloqueio ou nocaute gênico de CCR5 possa desempenhar na atenuação, progressão e até mesmo na cura da infecção causada pelo HIV.
Palavras Chave: HIV, CCR5, mutação, receptores.
ABSTRACT
Discovered in 1996, the CCR5 gene mutation proved capable of conferring resistance to HIV infection in the population. This led to intense research in an attempt to use this discovery as the basis for a curative therapy for HIV, given that most cells infected by the HIV virus express the CCR5 receptor on their surface. In this sense, this scientific article of systematic review seeks to analyze the available studies that investigated the use of therapies based on the destruction or blocking of the CCR5 receptor as a possible cure for HIV. The discovery of the role of the CCR5 gene and the protective effect of the mutation aroused a growing interest in the therapeutic role that the possible blockade or gene knockout of CCR5 could play in the attenuation, progression and even in the cure of the infection caused by HIV.
Keywords: HIV, CCR5, mutation, receptors.
INTRODUÇÃO
O HIV é uma doença infecciosa causada pelo vírus da imunodeficiência humana (HIV) que afeta milhões de pessoas em todo o mundo. Desde sua descoberta, em meados da década de 1980, o HIV se tornou uma das principais ameaças à saúde pública global, causando milhões de mortes e comprometendo o bem-estar de milhões de pessoas. Embora a terapia antirretroviral (TARV) tenha transformado o tratamento do HIV, permitindo que as pessoas que vivem com o vírus possam levar vidas mais longas e saudáveis, ainda não há cura para a doença. Por esse motivo, a pesquisa em busca de uma cura para o HIV continua sendo uma prioridade para a comunidade científica (COHEN, 2004).
Em 1996, foi descoberta uma mutação no gene CCR5 que se mostrou capaz de conferir resistência à infecção pelo HIV em cerca de 1% da população europeia. A mutação CCR5-Δ32 elimina completamente a expressão do receptor CCR5, que é usado pelo HIV para entrar nas células humanas. Isso levou a uma intensa pesquisa na tentativa de utilizar essa descoberta como base para uma terapia curativa para o HIV, tendo em vista que a maioria das células infectadas pelo vírus HIV expressam o receptor CCR5 em sua superfície (GOLDANI, 2008).
Desde então, diversos estudos foram conduzidos para investigar a possibilidade de utilizar terapias baseadas na destruição ou bloqueio do receptor CCR5 como uma possível cura para o HIV. Essas abordagens terapêuticas visam impedir a entrada do HIV nas células humanas, privando o vírus de um dos seus principais alvos.
Este artigo científico de revisão sistemática tem como objetivo analisar os estudos disponíveis que investigaram o uso de terapias baseadas na destruição ou bloqueio do receptor CCR5 como uma possível cura para o HIV. Serão examinados os resultados desses estudos, bem como as limitações e desafios associados a essas abordagens terapêuticas.
Em suma, esta revisão sistemática fornecerá uma análise abrangente dos estudos disponíveis sobre o uso de terapias baseadas na destruição ou bloqueio do receptor CCR5 como uma possível cura para o HIV. Embora a descoberta da mutação CCR5-Δ32 tenha levado à esperança de que a destruição do receptor CCR5 pudesse ser a chave para a cura do HIV, ainda há muito a ser entendido sobre as implicações clínicas e os possíveis efeitos colaterais dessas abordagens terapêuticas.
2. METODOLOGIA
Trata-se de uma revisão sistemática de literatura direcionada para estudos brasileiros e internacionais que abordam a mutação no receptor CCR5 e a resistência ao HIV e será guiada pela questão de pesquisa: “será que a destruição desses receptores é a cura para essa doença?”. O estudo obedecerá aos seguintes critérios de inclusão: estudos que abordem a mutação no receptor CCR5 e a resistência ao HIV, que permitam acesso aos textos completos.
2.1 Procedimento de coleta e análise dos dados
A coleta de dados irá se constituir a partir de estudos publicados em base de dados eletrônicas: Lilacs (Literatura Latino-americana e do Caribe em Ciências da Saúde), Web of Science, nos portais CAPES e BVS (Biblioteca Virtual em Saúde) e na biblioteca PubMed (National Library of Medicine), e ocorrerá com busca ativa iniciada em março de 2023 com o auxílio dos seguintes descritores: ”Mutação no receptor CCR5”, “Resistência ao HIV”, “CCR5 e a cura para o HIV”. Um instrumento de coleta de dados do tipo formulário será desenvolvido, o qual conterá os seguintes itens: Ano de publicação, autores, principais resultados e conclusão, visando a extração e organização de informações necessárias para que se possa alcançar os objetivos propostos para essa pesquisa.
Posterior a busca eletrônica será avaliado se os títulos dos estudos se adequam aos objetivos do projeto, após, será feita a leitura dos resumos para que por conseguinte seja feita a leitura dos artigos na íntegra. Os estudos passarão por uma avaliação de qualidade metodológica através da escala Assessment of Multiple Systematic Reviews (AMSTAR), instrumento que busca evidências de validade e confiabilidade de determinadas pesquisas. Os artigos que atenderem aos critérios de elegibilidade, irão compor um quadro de origem autoral que evidenciará os resultados e sua confiabilidade.
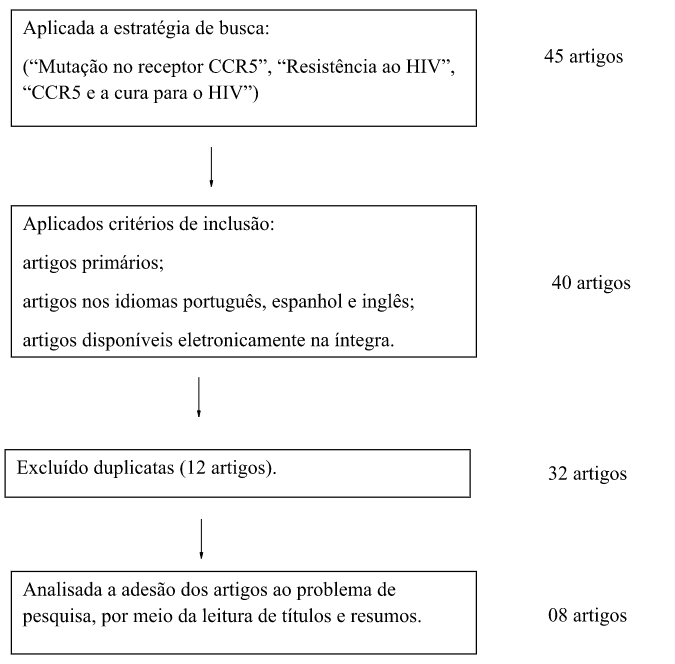
3.RESULTADOS
Foram encontrados 45 artigos com o tema pesquisado, sendo que, após os critérios de inclusão e exclusão de duplicatas, restaram 07 artigos que foram utilizados no presente trabalho.
Figura 1 – Seleção dos artigos. São Paulo, 2023.
4. REFERENCIAL TEÓRICO
4.1 Vírus da imunodeficiência humana do tipo 1 (hiv-1)
Existem duas variantes do HIV, o HIV-1 e o HIV-2. O HIV-1 é mais prevalente e patogênico, sendo responsável pela vasta pandemia global. Estudos filogenéticos e epidemiológicos sugerem que o HIV-1 possivelmente evoluiu a partir do vírus da imunodeficiência símia (SIVcpz) do chimpanzé, com o qual está intimamente relacionado. A transmissão do vírus entre espécies provavelmente ocorreu por meio do contato humano com primatas, especificamente no contexto da caça, quando humanos consumiam a carne de chimpanzé contaminada com o vírus (COHEN, 2004).
O HIV é um retrovírus que pertence à família Lentiviridae, com um genoma de 9 kb que contém nove genes que codificam quinze proteínas. Estruturalmente, o HIV é composto por um envelope externo, que contém as glicoproteínas de superfície gp120 e gp41, além de uma camada interna que contém uma membrana lipídica, uma matriz proteica e um capsídeo cônico que envolve dois filamentos simples de RNA, ligados a proteínas que formam o nucleocapsídeo. O vírus também contém três enzimas essenciais para a transcrição e replicação viral: transcriptase reversa, protease e integrase (SANTOS. 2020).
O envelope do HIV é formado por uma bicamada lipídica derivada da membrana da célula hospedeira e é composto por glicoproteínas de superfície gp120, que estão ancoradas ao vírus por meio de interações com a glicoproteína transmembrana gp41. As enzimas integrase, protease e transcriptase reversa são componentes internos essenciais para a transcrição e replicação viral. O HIV é um vírus de RNA e seu material genético está envolvido por uma estrutura proteica conhecida como capsídeo.
Em 1983, um grupo de pesquisadores liderados por Luc Montaignier do Instituto Pasteur de Paris, França, isolou pela primeira vez o Vírus da Imunodeficiência Humana tipo 1 (VIH-1 ou HIV-1, do inglês Human Immunodeficiency Virus), denominando-o de LAV (Lymphadenopathy Associated Virus ou Vírus Associado à Linfadenopatia) (BARRÉ-SINOUSSI et al., 1983).
Em 1984, a equipe liderada pelo pesquisador Dr. Robert Gallo nos Estados Unidos, isolou o vírus e denominou-o de HTLV-III (Vírus Linfotrópico de Células T-Humanas do tipo III). Em 1986, a Organização Mundial da Saúde recomendou o uso do termo HIV (Vírus da Imunodeficiência Humana) para referir-se ao vírus causador da Síndrome da Imunodeficiência Adquirida (AIDS). A partir de então, esforços foram direcionados para o desenvolvimento de medicamentos e vacinas para combater a doença (MINISTÉRIO DA SAÚDE, 2010).
No Brasil, o Ministério da Saúde reportou o primeiro caso de AIDS em 1980, assim como o primeiro óbito relacionado à doença. Em 1988, uma equipe liderada pelo pesquisador Dr. Bernardo Galvão Castro Filho isolou o HIV-1 no país, representando um marco simbólico para a pesquisa brasileira (GALVÃO, 2002).
Em 2010, 1,8 milhão de pessoas morreram em decorrência da AIDS no mundo. No entanto, desde 1999, quando a epidemia atingiu seu pico globalmente, o número de novas infecções diminuiu cerca de 19%, incluindo os dados de novos infectados na África Subsaariana, que reduziu de 2,2 milhões em 2001 para 1,8 milhão em 2010 (UNAIDS, 2010). Essa tendência reflete uma combinação de fatores, incluindo o impacto dos esforços na prevenção da infecção pelo HIV-1 e o curso natural da epidemia.
A epidemia da infecção pelo HIV-1 e da AIDS é um fenômeno global, instável e dinâmico, cuja forma de ocorrência em diferentes regiões do mundo depende, dentre outros fatores, do comportamento humano. Como resultado das desigualdades sociais enfrentadas pela população brasileira, a propagação da infecção pelo HIV no país revela uma epidemia multifacetada, caracterizada principalmente pelos processos de heterossexualização, feminização, interiorização e pauperização em curso na sociedade brasileira (BRITO et al., 2001).
O HIV foi primeiramente relatado em 1981 nos Estados Unidos e esteve associado a indivíduos adultos de ambos os gêneros que apresentavam alguns aspectos em comum, como sarcoma de Kaposi, pneumonia por Pneumocystis carinii, comprometimento do sistema imune e comportamento sexual de risco. Logo após a identificação do HIV, as pesquisas se voltaram para a identificação do agente etiológico da síndrome da imunodeficiência adquirida (AIDS).
O comportamento sexual de risco foi o principal fator de risco associado à infecção pelo HIV-1 nos primeiros anos da epidemia de AIDS (1980 a 1986), com destaque para o sexo anal e vaginal sem proteção. Em seguida, entre 1987 a 1991, a epidemia passou a se caracterizar pela transmissão sanguínea, principalmente na subcategoria de uso de drogas injetáveis, particularmente em regiões como a Ásia e o Leste Europeu, dando início a um processo caracterizado pela pauperização e interiorização da epidemia. Outros modos de transmissão do vírus que passaram a contribuir para a disseminação do HIV-1 nesse período foram o uso de hemocomponentes e o compartilhamento de objetos cortantes e perfurantes, os quais diminuíram drasticamente na maioria dos países desenvolvidos com a implantação de testes sorológicos e outras medidas de prevenção nos bancos de sangue e serviços de saúde.
Ao longo dos últimos 20 anos, um grande aumento na quantidade de casos por exposição sexual heterossexual vem sendo observado. Este fato reflete o número de milhões de mulheres e homens vivendo com HIV no mundo, caracterizando a heterossexualização da epidemia do HIV/AIDS. A transmissão sexual, incluindo a vertical, representa atualmente a forma predominante de transmissão do HIV no mundo. Particularmente, a transmissão do HIV de mãe para filho responde pela maioria dos casos pediátricos de AIDS. Esse tipo de transmissão ocorre com maior frequência durante o parto e o período de amamentação. Medidas de prevenção como o uso de antirretrovirais durante a gravidez e o parto, o parto por cesárea e a proibição da amamentação em caso de infecção materna têm contribuído para reduzir a transmissão vertical do HIV (CEZAR, 2014).
4.2 Estrutura e classificação do HIV
O Vírus da Imunodeficiência Humana (HIV) é um membro da família Retroviridae, subfamília Lentivirinae, que apresenta uma grande variabilidade genética. Existem duas formas de HIV que causam a AIDS, o HIV-1 e o HIV-2. O HIV-1 é subdividido em três grupos (M, N e O), sendo o grupo M predominante composto por 11 subtipos. Já o HIV-2 abrange seis diferentes linhagens filogenéticas, indo do subtipo A até o subtipo F (HAHN et al., 2000).
A estrutura do HIV-1 é composta por um nucleocapsídeo central de 100 nm de diâmetro, cercado por um envelope circular, constituído pela proteína de superfície extracelular gp120 e a proteína transmembranar gp41. O core interno é formado por três tipos de proteínas: p24 (capsídeo), p6 e p7 (nucleocapsídeo) e p17 (matriz). O genoma viral é composto por duas fitas de RNA de 9,2 kb cada, associadas a outras proteínas importantes, como a transcriptase reversa (p24), a integrase (p32) e a protease (p10) que desempenham funções catalíticas no processo de replicação viral (WATTS et al., 2009).
Após a infecção, o HIV-1 se replica dentro da célula hospedeira, usando a enzima transcriptase reversa para converter seu RNA em uma fita dupla de DNA. Esse DNA viral é incorporado ao DNA da célula hospedeira, formando o provírus. No entanto, devido às altas taxas de erro da transcriptase reversa e à dinâmica da replicação viral, o HIV-1 apresenta uma marcante variabilidade genotípica, que é agravada pela sua propriedade de recombinação e pela ausência de um sistema de correção de erros (ROBERTS et al., 1988).
Isso resulta em praticamente cada partícula viral contendo um genoma distinto dos demais. Tal elevada taxa de variação é responsável pelo surgimento de cepas virais com características biológicas distintas (SIMMONDS et al., 1990) e/ou resistentes à terapia anti-retroviral (COFFIN, 1995). Como resultado, essa grande variabilidade genética do vírus é um dos maiores desafios no desenvolvimento de uma vacina universal (COFFIN, 1995).
O HIV-1 tem um genoma composto por três genes necessários para sua replicação (gag, env e pol) e outros genes adicionais que regulam sua expressão e são importantes na patogênese viral (tat, nef, rev, vif, vpr e vpu). O gene codifica as glicoproteínas de superfície e transmembrana do envelope viral (gp120 e gp41), que reconhecem e penetram a célula-hospedeira. O gene gag codifica as proteínas do capsídeo e matriz protéica viral (p17 e p24), enquanto o gene pol codifica a transcriptase reversa, protease e integrase, responsáveis pela conversão do RNA em DNA, maturação protéica dos componentes virais e integração do material genético viral ao DNA da célula infectada.
Os outros genes adicionais regulam a transcrição (tat), expressão do RNA mensageiro (rev), infectividade (nef), formação do complexo de integração (vif), localização nuclear desse complexo (vpr) e liberação viral (vpu). As extremidades do genoma contém duas regiões repetidas, chamadas LTR (do inglês Long Terminal Repeat), cujas sequências são essenciais para a integração do DNA proviral no genoma do hospedeiro e também para a regulação da transcrição do genoma do HIV-1 por meio da ligação dos fatores de transcrição, como NFκB (WATTS et al., 2009).
4.2 Co Receptores do HIV
Conforme mencionado anteriormente, a infecção pelo HIV-1 em células depende de uma série de interações entre a subunidade de superfície (gp120) da glicoproteína do envelope viral (env), o receptor celular (molécula CD4) e os receptores de quimiocinas (CCR5 ou CXCR4, principalmente), que agem como co receptores em células-alvo (CHOE et al., 2003; NAZARI & JOSHI, 2008; GORRY & ANCUTA, 2010).
Quimiocinas (abreviação de citocinas quimiotáticas) são pequenos polipeptídeos de cerca de 8-12 kDa, que contêm duas pontes dissulfeto internas e desempenham um papel importante na regulação funcional do sistema imunológico, estimulando, por exemplo, a migração de diversas subpopulações de leucócitos. Algumas quimiocinas podem ser produzidas por várias células em resposta a estímulos inflamatórios e recrutam leucócitos para os locais de inflamação. Outras quimiocinas, entretanto, são produzidas normalmente em vários tecidos e recrutam leucócitos para esses tecidos mesmo na ausência de inflamação.
As quimiocinas são classificadas em famílias com base no número e na localização dos resíduos de cisteína N-terminal. As duas principais famílias são a das quimiocinas CC, nas quais os resíduos de cisteína são adjacentes, e a das quimiocinas CXC, nas quais esses resíduos são separados por um aminoácido (ABBAS & LICHTMAN, 2005). Alguns linfócitos T ativados secretam quimiocinas, como RANTES, MIP-1α, MIP-1β e CCL-5, que diminuem a infecção por algumas cepas do HIV-1, pois competem com o vírus pelo mesmo receptor, o CCR5, para se ligarem na célula (COCHI et al., 1995). Onze receptores diferentes para quimiocinas CC (chamados CCR1 a CCR11) e seis para quimiocinas CXC (chamados CXCR1 a CXCR6) já foram identificados.
Todos os receptores de quimiocinas exibem uma estrutura característica com sete domínios α-helicoidais transmembrânicos. Tais receptores possuem sobreposição de especificidades para as quimiocinas dentro de cada subfamília e o padrão de expressão celular desses receptores determina quais os tipos celulares que respondem a cada quimiocina. Certos receptores de quimiocinas, especialmente CCR5 e CXCR4, atuam como co receptores para o HIV-1 (ABBAS & LICHTMAN, 2005).
Os receptores CCR5 e CXCR4 são expressos em células T CD4+ e macrófagos. O CCR5 atua como receptor natural para β-quimiocinas, incluindo RANTES, MIP-1α, MIP-1β e CCL-5, enquanto o CXCR4 é receptor para α-quimiocinas, como SDF-1α. Embora menos comuns, outras moléculas, como CCR2b, CCR3, CCR8, CCR9 e CXCR6, também podem ser usadas por algumas cepas de HIV-1 para infectar as células, mesmo na ausência de CCR5 ou CXCR4.
As cepas do HIV-1 que usam o CCR5, chamadas cepas R5, são responsáveis pela maioria das novas infecções e predominam nas fases iniciais e crônicas da infecção pelo HIV-1. Por outro lado, as cepas do vírus que usam o CXCR4, chamadas cepas X4, surgem na infecção tardia e contribuem para a progressão mais rápida da doença. Existem também as cepas R5/X4, que possuem tropismo para os dois co receptores. A glicoproteína de superfície gp120 do vírus interage com a região amino-terminal (N-terminal) e com o segundo loop extracelular (ECL2) da CCR5. Enquanto a região do envelope viral das cepas X4 interage com o primeiro e o segundo loop extracelular (ECL1 e ECL2, respectivamente) da molécula CXCR4. As cepas R5/X4 podem interagir tanto com a região N-terminal da CCR5 quanto com o ECL1 e o ECL2 da CXCR4. No entanto, o CXCR4 é preferencialmente usado pelas cepas R5/X4 para entrar nas células (COHEN, 2004).
Em geral, cerca de 4 a 5 anos após a infecção inicial pelo subtipo B do HIV-1, mais de 50% dos indivíduos infectados apresentam uma evolução das cepas R5 para cepas X4 do vírus, o que está correlacionado com um declínio rápido das células T-CD4+ e a progressão para a AIDS. Os mecanismos moleculares que levam à troca do receptor utilizado pelo HIV-1 ainda não estão completamente compreendidos. No entanto, alguns estudos indicam que ocorre uma redução no fitness viral das cepas R5, o que pode permitir o surgimento dos vírus R5/X4. Para evoluir para as cepas X4, as cepas R5 do HIV-1 sofrem mutações na alça V3 (terceira alça variável) da gp120, além de mudanças na eficiência com que a proteína env se liga ao co receptor CCR5 (COETZER et al., 2008).
As mudanças no tropismo viral para células T-CD4+ têm um grande impacto na patogênese do HIV-1. Além disso, há um interesse crescente no uso dos co receptores apresentados por diferentes cepas do HIV-1, devido ao desenvolvimento de uma nova classe de drogas antirretrovirais, os antagonistas dos correceptores ou bloqueadores/inibidores de fusão. Essas drogas podem bloquear tanto os co receptores CCR5 quanto CXCR4, bem como a molécula CD4, impedindo a fusão do HIV-1 com a membrana celular (SIERRA et al., 2007).
4.2.1 Coreceptor CCR5
O gene CCR5 está localizado na posição 21 do braço curto do cromossomo 3 (3p21) e codifica o receptor de quimiocinas CC humano do tipo 5, CCR5. Ele é composto por 4 éxons e 2 íntrons, e não há íntron entre os éxons 2 e 3. A transcrição é iniciada em 2 diferentes promotores, ambos ricos em adenina e timina. Padrões de splicing alternativo podem gerar dois transcritos diferentes da CCR5: a CCR5-A e a CCR5-B, que codificam uma proteína idêntica (CCR5) em diferentes tipos celulares. O padrão de regulação da expressão desses transcritos varia em resposta a uma variedade de sinais extracelulares, como citocinas e quimiocinas (MUMMID et al., 1997).
A proteína CCR5 é um membro da família de receptores de β-quimiocinas e apresenta sete domínios transmembranares anfipáticos, semelhante aos receptores acoplados à proteína G (GPCRs) (FEDERSPPIEL et al., 1993; HERZOG et al., 1993; NOMURA et al., 1993). A CCR5 é composta por 352 aminoácidos e tem 40,6 kDa, com um domínio amino-terminal (N-Terminal) voltado para a superfície extracelular e um domínio carboxi-terminal (C-Terminal) voltado para o citoplasma da célula. Ela sofre modificações pós-traducionais, como sulfatação de tirosinas adjacentes a aminoácidos ácidos e O-glicosilação no domínio N-Terminal, que favorecem sua interação com seus ligantes, como as β-quimiocinas e o HIV1 (FARZAN et al., 2000; CORMIER et al., 2000). A CCR5 também tem 3 loops extracelulares (ECL1, ECL2 e ECL3) e 3 loops intracelulares (ICL1, ICL2 e ICL3), sendo que o segundo loop intracelular (ICL2) contém o domínio de ligação à proteína G.
A CCR5 é expressa na superfície de células T, macrófagos, monócitos, Células Dendríticas do tipo DC-SIGN e Células de Langerhans (CARRINGTON et al., 1997; NAZARI & JOSHI, 2008). Sua expressão é regulada a curto prazo, em nível proteico, por mecanismos como internalização, sequestro e dessensibilização, e a longo prazo, pela taxa de transcrição do gene e estabilidade do RNA mensageiro (MUMMID et al., 1997). Os ligantes para este receptor incluem as β-quimiocinas MCP-2, MIP-1 α, MIP-1 β e RANTES, e sua ligação causa a migração de leucócitos para os sítios de inflamação (CARRINGTON et al., 1997)
Uma segunda classe de ligantes para a CCR5 inclui as cepas R5 do HIV-1, que se ligam à gp210. Neste contexto, a proteína CCR5 atua como receptor para o vírus (DRAGIC et al., 1996; DENG et al., 1996). Isso pode ser confirmado pelo fato de que quimiocinas como MIP-1α, MIP-1β e RANTES inibem a infecção de células T CD4+ por cepas R5 do HIV-1 no estágio de entrada do vírus, bloqueando a fusão célula-célula mediada por env, competindo pela mesma proteína, CCR5, na célula (DRAGIC et al., 1996).
Em relação ao papel da CCR5 como co receptor para o HIV-1, sabe-se que determinadas regiões ou domínios da molécula CCR5 são importantes na interação com a gp120 viral (ATCHISON et al., 1996; RUCKER et al., 1996). Mais especificamente, a glicoproteína de superfície gp120 do vírus interage com a região amino-terminal (N-terminal), entre os resíduos 2 e 18, e com o segundo loop extracelular (ECL2) da CCR5. Alterações nestes domínios podem interferir nos processos de ligação entre a CCR5 e a gp120, bem como na fusão da membrana do HIV-1 com a membrana celular, afetando a entrada do vírus na célula (FARZAN et al., 2000).
Por este motivo, indivíduos com padrão homozigoto mutante no gene da CCR5 (genótipo ∆32/∆32) para uma deleção de 32 pares de bases, compreendida entre os nucleotídeos 794 e 825 na região do gene que codifica o segundo loop extracelular da proteína CCR5, possuem resistência contra a infecção por cepas R5 do HIV-1, pelo menos por via sexual. Isso corrobora o fato de que a CCR5 é o principal receptor utilizado por cepas primárias do HIV-1 (SAMSON et al., 1996). Além disso, como as cepas R5 predominam durante a infecção primária do HIV-1, a CCR5 tornou-se um alvo atraente para o desenvolvimento de terapias contra a infecção por este vírus. Diferentes mecanismos têm sido elaborados para bloquear sua função como receptor do HIV-1 ou diminuir sua expressão na superfície da célula-alvo.
A terapia gênica, por exemplo, utiliza intra-quimiocinas (quimiocinas intracelulares que se ligam aos receptores de quimiocinas), para prevenir a expressão da CCR5 na superfície da célula; intra anticorpos (fragmentos de anticorpos intracelulares) contra a região N-Terminal da CCR5, para diminuir a expressão de CCR5; e ainda pequenos RNAs de interferência (siRNA) e RNA antisense, que diminuem a síntese do co receptor (NAZARI & JOSHI, 2008).
5. RESULTADOS E DISCUSSÃO
De acordo com pesquisa realizada por Deeks (2015), desde o início dos estudos sobre o HIV, o receptor CD4 encontrado na superfície das células CD4 foi considerado crucial para a entrada do vírus nessas células. Contudo, pesquisadores descobriram, na década de 90, que além do receptor CD4, as proteínas CCR5 e CXCR4, também presentes na superfície das células CD4, atuam como co-receptores do HIV. Nos estágios iniciais da infecção, o HIV-1 tem preferência pelo co receptor CCR5 (HIV R5-Trópico), enquanto as cepas virais que possuem tropismo pelo co receptor CXCR4 (HIV X4-Trópico) aparecem com mais frequência nos estágios tardios da infecção. Existem ainda cepas que têm tropismo por ambos os correceptores, denominadas cepas R5/X4. Os co receptores são essenciais para a primeira etapa do mecanismo de infecção do HIV na célula CD4, que é a ligação e entrada do vírus. Essa etapa é caracterizada pela ligação da proteína gp120, presente na superfície do vírus, às células CD4 por meio do receptor CD4, o que desencadeia uma mudança conformacional em gp120 e a ligação do complexo ativo das glicoproteínas virais gp120 e gp41 ao co receptor CCR5 ou CXCR4.
Em seguida, ocorre a fusão da membrana viral com a membrana da célula CD4, e o vírus libera no citoplasma da célula o seu RNA e as enzimas transcriptase reversa, integrase e protege. Na segunda etapa, ocorre a transcrição reversa do RNA viral em DNA pela enzima transcriptase reversa e, na terceira etapa, ocorre a integração do DNA viral ao genoma da célula hospedeira pela enzima integrase. Posteriormente, na quarta etapa, ocorre a replicação viral, em que novas moléculas de RNA mensageiro do vírus são transcritas pela maquinaria da célula hospedeira, e as suas proteínas são traduzidas e translocadas para a superfície celular. Na quinta etapa, ocorre o processo de montagem da estrutura central viral, em que longas cadeias de proteínas sintetizadas são cortadas em proteínas individuais pela enzima protease. Assim, novos vírus são formados e, por fim, na sexta etapa, ocorre a maturação viral. Uma vez maduros, os novos vírus nascentes estão prontos para infectar outras células CD4.
Liu et ai. (1996) observaram inicialmente que alguns indivíduos expostos ao HIV-1 não eram infectados e, além disso, suas células T CD4+ eram altamente resistentes à entrada do vírus in vitro. Estudos posteriores demonstraram que essa resistência poderia ser causada por uma mutação no gene que codifica o CCR5. Uma deleção de 32 pares de bases, conhecida como mutação delta 32 do CCR5 ou CCR5Δ32, causa um frameshift, ou seja, uma mudança no frame de leitura durante a tradução, afetando as três últimas regiões transmembranares do CCR5, especificamente no segundo loop extracelular onde a proteína Ocorre o sítio de ligação G responsável pela transdução do sinal celular. O alelo mutante contém 215 aminoácidos, enquanto o alelo selvagem possui 352 aminoácidos, gerando uma proteína truncada que não pode ser detectada na superfície celular. (DEAN et al., 1996; SAMSON et al., 1996; LIU et al., 1996).
Estudos demonstram que o alelo delta 32 surgiu de uma mutação simples e que esse evento tem origem recente no nordeste da Europa (LIBERT et al., 1998). Portanto, a frequência na população caucasiana é maior do que em outros grupos étnicos, em torno de 10%. Essa mutação é autossômica recessiva, portanto, indivíduos heterozigotos para CCR5 delta 32 ainda podem expressar receptores de forma diminuída, enquanto indivíduos homozigotos para a mutação carecem de receptores CCR5 (SAMSON et al., 1996).
Esses fatos constituem evidência da capacidade duradoura de controle do vírus. Uma das hipóteses para um indivíduo ter o perfil de um progresso lento ou controlador de elite é a mutação delta 32 do CCR5, pois é fundamental no momento da interação vírus-célula hospedeira que haja uma conexão com este co-receptor. Portanto, na sua ausência ou baixa expressão, a entrada do vírus no meio intracelular fica prejudicada (CARRINGTON et al., 1999). No Brasil, a frequência de heterozigose varia de 6,8% a 14,2% na população caucasiana; de 1% para 6,4% na população negra e de 0,4% para 2,2% na população indígena (BOLDT et al., 2009; VARGAS et al., 2006; CARVALHO et al., 2004; CARVALHARES et al., 2005).
Vangelista (2018) reportou que a identificação do CCR5 como co-receptor do HIV na célula CD4 permitiu a descoberta de que ele é um alvo importante para o desenvolvimento de uma nova classe de drogas antirretrovirais, conhecida como antagonistas de co-receptores ou bloqueadores/inibidores de interação. Um exemplo disso é o medicamento Maraviroc (aprovado para uso pela ANVISA em 2007), uma pequena molécula que se liga ao co-receptor CCR5 e induz uma mudança em sua conformação estrutural, especificamente nos sítios de interação do HIV, levando ao bloqueio da entrada do vírus na célula CD4. No entanto, a eficácia do Maraviroc é limitada, visto que algumas cepas virais conseguem usar alternativamente o co-receptor CXCR4, enquanto outras cepas são capazes de interagir com o CCR5 mesmo quando está ligado ao medicamento. Além disso, há evidências de toxicidade, causada pela falta de especificidade para o receptor. Outros antagonistas do co-receptor CCR5 foram desenvolvidos, mas raramente são utilizados por serem de alto custo.
A deleção de 32 pares de bases no gene CCR5 (mutação ∆32) resulta em uma alteração no quadro de leitura durante a transcrição do gene e na terminação prematura da tradução do transcrito, após o aminoácido 174, afetando as últimas três regiões transmembranares da CCR5 e o sítio de ligação da proteína G, responsável pela transdução de sinais extracelulares (SAMSON et al., 1996). A produção de uma proteína truncada, que não permite a fusão de células T CD4+ com células que expressam a gp120 viral e pode não ancorar adequadamente na membrana da célula, é responsável pela resistência à infecção pelo HIV-1 em indivíduos que apresentam essa mutação em homozigose (LIU et al., 1996; SAMSON et al., 1996). Já indivíduos que possuem essa mutação em heterozigose (CCR5/∆32) parecem ser parcialmente resistentes à infecção, apresentando progressão mais lenta para a AIDS, quando comparados aos homozigotos selvagens (CCR5/CCR5) (DENG et al., 1996).
A resistência parcial atribuída à heterozigose é consequência da diminuição da expressão da proteína CCR5 na membrana celular em indivíduos heterozigotos (LITTMAN, 1998). Na América do Sul, incluindo o Brasil, a frequência do alelo CCR5-∆32 varia de 0 a 6,5% (GRIMALDI et al., 2002). Na Bahia, a prevalência do genótipo heterozigoto (CCR5/∆32) foi de 5,3% em indivíduos não infectados (GRIMALDI et al., 2002). No entanto, o alelo CCR5-∆32 não é o único fator responsável pela modulação da infecção pelo HIV-1 através do co receptor CCR5.
Até o momento, foram descritas 25 mutações diferentes no gene CCR5 (CARRINGTON et al., 1999), as quais apresentam distribuição geográfica e étnica diferentes (O’BRIEN et al., 2000). Entre essas mutações, existem genótipos e/ou haplótipos que conferem resistência à infecção pelo HIV-1. A mutação p.C101X, por exemplo, cria um códon de parada antes do terceiro domínio transmembranar da CCR5, gerando uma proteína incapaz de mediar a entrada do HIV-1 na célula devido à sua expressão diminuída na superfície celular, com efeito semelhante à mutação ∆32. Assim, indivíduos homozigotos para essa mutação são resistentes à infecção pelo HIV-1 (BLANPAIN et al., 2000).
Além das mutações identificadas na região codificante, algumas mutações no promotor do gene CCR5 podem interferir no curso clínico da AIDS, acelerando ou retardando a progressão da doença em indivíduos infectados pelo HIV-1, mesmo que não afetem a infecção por cepas R5 do vírus (MARTIN et al., 1998). Essas mutações podem comprometer a regulação gênica, afetando a transcrição e/ou a expressão da proteína na superfície celular e provavelmente afetando a patogenicidade do HIV-1 (McDERMOTT et al., 1998).
Algumas mutações descritas nessa região do gene, como -2733A>G (29 A/G) e -2459A>G (303 A/G), estão associadas à aceleração da progressão da doença, devido ao aumento do nível de expressão da proteína CCR5 na membrana (McDERMOTT et al., 1998; CLEGG et al., 2000; OMETTO et al., 2001; BRUMME et al., 1999; ROMAN et al., 2002). Por outro lado, as variações com efeito contrário levam a uma progressão mais lenta da doença, como a mutação -1835C>T (927 C/T) (BRUMME et al., 2001; LEWANDOWSKA et al., 2002).
Conforme mencionado anteriormente, a proteína CCR5 passa por modificações pós-traducionais, como a sulfatação de resíduos de tirosina no domínio N-terminal, que favorece sua ligação às quimiocinas MIP1-α e MIP1-β, bem como facilita a entrada do HIV-1 na célula hospedeira (CORMIER et al., 2000; FARZAN et al., 2000; HOFFMAN et al., 2002; SEIBERT et al., 2002). A região compreendida entre os resíduos 2 e 18 do domínio N-terminal da CCR5 constitui o sítio específico de interação entre a alça V3 da gp120 do HIV-1 e a CCR5. A sulfatação das tirosinas nos aminoácidos 3, 10, 14 e 15 é importante para a interação entre esse co-receptor e a proteína viral, e a perda de um ou mais desses grupamentos sulfato pode inibir a fusão do vírus com a célula-alvo (FARZAN et al., 2000).
Mutações no gene CCR5 que interferem na produção dos resíduos de tirosina do domínio N-terminal da proteína podem estar relacionadas à habilidade da CCR5 em se ligar à gp120. No entanto, novos estudos ainda questionam a possibilidade desses sítios específicos não serem os únicos responsáveis pela interação entre o co-receptor e o vírus (CONNIE et al., 2009). Alguns autores acreditam que a associação inicial entre a CCR5 e a gp120 é mediada pelo domínio N-terminal deste co-receptor, enquanto a maior especificidade da interação é mediada pelos demais domínios extracelulares da CCR5, principalmente o ECL2 (GERARD & GERARD, 1996; MONTECLARO & CHARO, 1996).
O domínio carboxi-terminal da CCR5 não interage diretamente com a gp120 do HIV-1, pois está localizado no meio intracelular. No entanto, outras modificações pós-traducionais, como a N-glicosilação (adição de um poli ou oligossacarídeo em um nitrogênio da cadeia lateral de resíduos de asparagina ou arginina) e a O-glicosilação da CCR5 na região C-terminal, também podem influenciar a entrada do HIV-1 em suas células-alvo. Alterações nos sítios de glicosilação podem ter consequências dramáticas para uma proteína, pois afetam seu dobramento e sua conformação, interferindo em partes distantes da mesma. Portanto, a perda de sítios de glicosilação no domínio C-terminal da CCR5 pode interferir na inserção da proteína na membrana celular, gerando uma proteína que sofreu uma conformação atípica (SLATER-HANDSHY et al., 2004).
Além disso, foi demonstrado que elementos críticos para a interação com o HIV-1, que funcionam como peptídeos imunogênicos, estão localizados na segunda alça extracelular (ECL2) do receptor. Algumas mutações na ECL2 podem comprometer a eficiência da entrada do HIV-1 na célula-alvo, levando a uma diminuição na progressão para AIDS (GERARD & GERARD, 1996; MONTECLARO & CHARO, 1996). Até o momento, não há dados sobre essas mutações nas regiões do gene CCR5 que codificam para os domínios N-Terminal, C-Terminal e ECL2 da proteína na população brasileira. Portanto, diante da realidade da infecção pelo HIV-1 e da AIDS no Brasil, essas informações podem ser importantes para o estudo do perfil epidemiológico da infecção pelo HIV-1 em nosso país.
A mutação CCR5∆32 é caracterizada pela deleção de 32 pares de bases que abrange o nucleotídeo 794 ao 825 na região de quadro de leitura aberto (ORF) do gene. Essa deleção gera uma mudança na matriz de leitura (também conhecida como mutação frameshift ou de mudança de quadro de leitura) durante a tradução, resultando na inclusão de sete novos aminoácidos após o aminoácido 174 da proteína e um término prematuro devido à formação de um códon de parada após o aminoácido 182. Dessa forma, o alelo mutado codifica uma proteína truncada que contém 215 aminoácidos, em comparação com a proteína selvagem que contém 352 aminoácidos.
A região afetada corresponde à ECL2 da proteína, que perde os três últimos domínios transmembranares, bem como regiões importantes de interação com a proteína G, responsável pela transdução de sinais extracelulares. Além da proteína CCR5 mutante não ser funcional, a mutação também afeta o local onde CCR5 é expressa nas células CD4, passando a ser localizada no citoplasma em vez de na superfície da célula, como ocorre com a proteína CCR5 selvagem. Assim, o fenótipo mutante do receptor CCR5 confere proteção contra a infecção causada pelo HIV, uma vez que esse vírus não consegue se ligar e entrar na célula CD4 do hospedeiro (CEZAR, 2014).
Esses estudos científicos também evidenciam que, embora a mutação CCR5∆32/∆32 proporcione proteção contra o HIV, portadores dessa mutação podem ter um pior prognóstico em casos de doenças inflamatórias. Ademais, a mutação aumenta o risco de mortalidade em casos de infecção pelo vírus do Nilo ocidental e/ou da influenza, além de predispor a problemas ósseos. Embora a técnica de edição gênica CRISPR tenha o potencial de gerar mutações pontuais e específicas no DNA, é importante destacar que ela também pode gerar mutações indesejadas (chamadas off-targets) e causar danos graves ao organismo.
Assim, a utilização da técnica de CRISPR em humanos deve considerar duas premissas importantes: em primeiro lugar, o pesquisador deve estar ciente de todas as consequências da mutação gerada, tanto positivas quanto negativas. Em segundo lugar, é essencial garantir que não ocorram off-targets, a fim de evitar a ocorrência de mutações indesejadas. Portanto, embora a técnica CRISPR seja promissora, os cientistas destacam que, atualmente, é necessário estabelecer normas para seu uso e aplicação, regulamentando o que é permitido ou não, para garantir sua utilização de forma racional e segura.
CONCLUSÃO
A descoberta de que o gene CCR5 codifica um receptor na célula CD4, que é essencial para a infecção pelo HIV, trouxe importantes contribuições para o entendimento sobre a patogênese desse vírus. Ademais, a constatação de que indivíduos homozigotos para a mutação CCR5 ∆32 apresentam um fenótipo que confere proteção contra a infecção pelo HIV abre e continua abrindo perspectivas para novas possibilidades terapêuticas contra o vírus, seja por meio do desenvolvimento de novos fármacos, do transplante de medula a partir de doadores portadores da mutação CCR5∆32/∆32, ou até mesmo a possibilidade de terapia gênica. Desse modo, os estudos que abordam o gene CCR5 contribuem para o desenvolvimento de novas abordagens de tratamento e proteção contra a infecção pelo HIV.
REFERÊNCIAS
ABBAS, A. K. Propriedades gerais das respostas imunológicas IN: ABBAS, AK; LICHTMAN, AH Imunologia celular e molecular. 2005.
BARRÉ-SINOUSSI, Françoise et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science, v. 220, n. 4599, p. 868-871, 1983.
BRITO, Ana Maria de; CASTILHO, Euclides Ayres de; SZWARCWALD, Célia Landmann. AIDS e infecção pelo HIV no Brasil: uma epidemia multifacetada. Revista da sociedade brasileira de medicina tropical, v. 34, p. 207-217, 2001.
CEZAR, Vagner Mendes; DRAGANOV, Patricia Bover. A História e as Políticas Públicas do HIV no Brasil sob uma Visão Bioética. Ensaios e Ciência C Biológicas Agrárias e da Saúde, v. 18, n. 3, 2014.
CHOE, Hyeryun et al. Tyrosine sulfation of human antibodies contributes to recognition of the CCR5 binding region of HIV-1 gp120. Cell, v. 114, n. 2, p. 161-170, 2003.
COETZER, Mia et al. Evolution of CCR5 use before and during coreceptor switching. Journal of virology, v. 82, n. 23, p. 11758-11766, 2008.
COFFIN, John M. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science, v. 267, n. 5197, p. 483-489, 1995.
COHEN, Myron S. HIV and sexually transmitted diseases: lethal synergy. Topics in HIV medicine: a publication of the International AIDS Society, USA, v. 12, n. 4, p. 104-107, 2004.
DEEKS, S. G.; OVERBAUGH, HIV infection. Nature Review Disease Primers, v.1, p.15035, 2015.
FARZAN, Michael et al. A tyrosine-sulfated peptide based on the N terminus of CCR5 interacts with a CD4-enhanced epitope of the HIV-1 gp120 envelope glycoprotein and inhibits HIV-1 entry. Journal of Biological Chemistry, v. 275, n. 43, p. 33516-33521, 2000.
GALVÃO, Shindo N, Acosta AX, Dourado I, Brites C, de Melo Carvalho O, Brito I, BouHabib DC, Galvão-Castro B. Prevalence of the CCR5 Delta32 mutation in Brazilian populations and cell susceptibility to HIV-1 infection. Hum Genet. 111(1):102-4, 2002.
GOLDANI, Luciano Zubaran. Descoberta do HIV: o reconhecimento. Clinical and Biomedical Research, v. 28, n. 3, 2008.
GRIMALDI, Rogério et al. Study of the CCR5-m303 mutation in three different ethnic groups from Brazil. Genetics and Molecular Biology, v. 28, p. 214-217, 2005.
HAHN, Beatrice H. et al. AIDS as a zoonosis: scientific and public health implications. Science, v. 287, n. 5453, p. 607-614, 2000.
LIU, Rong et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell, v. 86, n. 3, p. 367-377, 1996.
MINISTÉRIO DA SAÚDE, Secretaria de Vigilância em Saúde, Departamento de DST, AIDS e Hepatites virais. Boletim epidemiológico – AIDS e DST. Em http://www.aids.gov.br/sites/default/files/anexos/publicacao/2010/45974/vers_o_final_15923 .pdf, acessado em 30 de mar de 2023. Brasil, Ministério da Saúde, 2010.
NAZARI, Reza; JOSHI, Sadhna. CCR5 as target for HIV-1 gene therapy. Current gene therapy, v. 8, n. 4, p. 264-272, 2008.
PROGRAMA COMUM DAS NAÇÕES UNIDAS SOBRE HIV / AIDS.; ORGANIZAÇÃO MUNDIAL DE SAÚDE. Relatório de 2008 sobre a epidemia global de AIDS. 2010
ROBERTS, John D.; BEBENEK, Katarzyna; KUNKEL, Thomas A. The accuracy of reverse transcriptase from HIV-1. Science, v. 242, n. 4882, p. 1171-1173, 1988.
SANTOS, Ana Cláudia Freitas et al. Perfil epidemiológico dos pacientes internados por HIV no Brasil. Revista eletrônica acervo saúde, n. 48, p. e3243-e3243, 2020.
SIERRA, Saleta et al. Genotypic coreceptor analysis. European journal of medical research, v. 12, n. 9, p. 453, 2007.
SLATER-HANDSHY, Tiffany et al. HCV E2 glycoprotein: mutagenesis of N-linked glycosylation sites and its effects on E2 expression and processing. Virology, v. 319, n. 1, p. 36-48, 2004.
UNAIDS and WHO. AIDS epidemic update 2009. Em http://www.unaids.org/en/dataanalysis/epidemiology/2009aidsepidemicupdate, acessado em 30 de mar de 2023. Geneva, UNAIDS, 2010.
VANGELISTA, L AND VENTO, S. The expan ding Therapeutic Perspective of CCR5 Blockade. Frontiers in Immunology, v.8, p.1981-1987, 2018.
WATTS, Joseph M. et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature, v. 460, n. 7256, p. 711-716, 2009.