REGISTRO DOI: 10.5281/zenodo.8051010
Maria Eduarda Wilvert;
Ariana Centa;
Maria Aparecida Marques Habermann;
João Paulo Assolini.
Resumo: A osteogênese imperfeita (OI) é uma doença rara, definida como uma desordem genética e heterogênea do tecido conjuntivo. É caracterizada principalmente por fragilidade óssea, que resulta em fraturas recorrentes e deformidades esqueléticas, manifestações que tornam a doença conhecida como “doença dos ossos de vidro”. As manifestações extraesqueléticas da OI incluem entre tantos, esclera azulada, doenças pulmonares, anormalidades auditivas e a dentinogênese imperfeita. Com o presente estudo, objetiva-se definir a osteogênese imperfeita e, identificar e descrever sua fisiopatologia, características clínicas, diagnóstico, tratamento e seu impacto na vida dos pacientes acometidos através de uma revisão narrativa.
Palavras-chaves: Doença genética; Doença Óssea Congênita; Desenvolvimento infantil; Colágeno
Abstract: Osteogenesis imperfecta (OI) is a rare disease, defined as a genetic and heterogeneous disorder of the connective tissue. It is identified mainly by bony individuals, which results in recurrent fractures and skeletal deformities, manifestations that make the disease known as “glass bone disease”. The extraskeletal manifestations of OI include, among many, bluish sclera, lung disease, auditory abnormalities, and dentinogenesis imperfecta. The present study aims to define osteogenesis imperfecta and identify and describe its pathophysiology, clinical characteristics, diagnosis, treatment and its impact on the lives of affected patients through a narrative review.
Keywords: Genetic disease; Congenital Bone Disease; Child Development; Collagen
Introdução
A osteogênese imperfeita (OI) é uma desordem genética e heterogênea do tecido conjuntivo. Suas manifestações clínicas primárias envolvem principalmente o esqueleto, e por isso, é conhecida como a doença do osso quebradiço ou, doença dos ossos de vidro. Indivíduos com OI apresentam baixa massa óssea e consequente fragilidade óssea, que resulta em maior suscetibilidade a fraturas e deformidades dos ossos. Entretanto, a doença apresenta também características secundárias que incluem a esclera azul, perda auditiva, dentinogênese imperfeita, comprometimento da função pulmonar e anormalidades da válvula cardíaca (MARINI et al., 2017).
Cerca de 85% a 90% dos casos de OI são causados por mutações autossômicas dominantes dos genes collagen type I alpha 1 chain (COL1A1) ou collagen type I alpha 2 chain (COL1A2) que codificam as cadeias α-1 e α-2 do colágeno tipo I, que é responsável pelas propriedades elásticas dos ossos e da pele, sendo o principal componente da matriz extracelular dessas estruturas (MEI; ZHANG; ZHANG, 2022). As causas menos frequentes abrangem as mutações autossômicas recessivas de genes envolvidos nas vias metabólicas do colágeno ou de proteínas associadas a cartilagem ou de mineralização óssea (MORABITO et al., 2022).
Devido a diversidade de manifestações clínicas da OI o seu manejo é complexo e individual. Indivíduos com OI necessitam de um atendimento amplo em saúde, alcançado por uma equipe multidisciplinar, uma vez que não há uma única especialidade capaz de atender a todas as necessidades dos pacientes com a doença (MARR; SEASMAN; BISHOP, 2017).
Estima-se que a prevalência da OI seja de 6 a 7 de cada 100.000 nascimentos, e que a sobrevida dos pacientes esteja relacionada aos fenótipos manifestados. Dessa maneira, pacientes com fenótipos mais leves possuem maior tempo de sobrevida em relação aqueles com a forma grave da doença (SPONER; KORBEL; KUCERA, 2022).
Independentemente da gravidade, o distúrbio apresenta diversos comprometimentos ao longo da vida que merecem o adequado diagnóstico e tratamento, sendo necessário dar continuidade aos estudos sobre essa desordem de pouca prevalência e elevada complexidade. Diante do exposto, objetiva-se com a presente revisão narrativa definir a OI, identificar e descrever sua fisiopatologia, características clínicas, diagnóstico, tratamento e seu impacto na vida dos pacientes acometidos.
Metodologia
Trata-se de um estudo de revisão narrativa sobre a fisiopatologia, classificação, manifestações clínicas, diagnóstico e tratamentos da OI. Para a identificação de estudos sobre o tema, foram utilizadas as bases PubMed (U.S. National Library of Medicine), a Biblioteca Virtual em Saúde (BVS) e SCIELO (Scientific Electronic Library Online). Os termos descritos a seguir foram selecionados nos campos título, resumo ou tópico, para a localização das publicações, associados aos operadores lógicos “AND”, para relacionar termos, e “OR”, para somar termos. Os termos de pesquisa utilizados para a investigação em todos as bases de dados foram: (”osteogênese imperfeita”) AND (“manifestações clínicas” OR “tratamento” OR “diagnóstico” OR “fisiopatologia” OR “classificações”) e, na língua inglesa (”osteogenesis imperfecta”) AND (“clinical manifestations” OR “treatment” OR “diagnosis” OR “pathophysiology” OR “classifications”). Além dos artigos, foram utilizados protocolos clínicos do Ministério da Saúde. As pesquisas das bases de dados foram limitadas a artigos em língua inglesa e portuguesa, publicados nos últimos 10 anos até abril de 2023.
Osteogênese Imperfeita (OI)
Define-se a osteogênese imperfeita (OI) como uma doença hereditária generalizada do tecido conjuntivo. Sua causa está relacionada principalmente com mutações dominantes nos genes COL1A1 ou COL1A2 que codificam as cadeias α-1 e α-2 da síntese do colágeno tipo I, proteína de maior abundância no corpo humano. Raramente, pode ser ocasionada por mutações recessivas dos genes relacionados ao colágeno. Ainda, as causas da OI podem ser divididas entre aquelas com defeito qualitativo ou quantitativo do processamento do colágeno (HALD et al., 2018).
Por se tratar de um grupo de desordens com manifestações esqueléticas caracterizadas por deformidade e fragilidade óssea, que levam a fraturas de membros e de vertebras, a OI é muitas vezes referida como a “doença dos ossos frágeis” ou “doença dos ossos de vidro”. Além dessas características primárias, a OI pode apresentar também fenótipos extraesqueléticos que envolvem a dentinogênese imperfeita, esclera azulada, comprometimento cardiopulmonar, frouxidão da pele e articulações. Devido a essa variabilidade de manifestações, a OI é considerada um distúrbio generalizado do tecido conjuntivo e não unicamente esquelético (GARIBALDI et al., 2022).
Ademais, salienta-se que as doenças raras são aquelas com grande variedade de sinais e sintomas que em indivíduos acometidos com a mesma condição manifestam-se de forma diferenciada. No Brasil são consideradas doenças raras aquelas que possuem uma prevalência de até 65 pessoas em cada 100.000 indivíduos (BRASIL, 2022). Nesse sentido, pode-se afirmar que a OI está dentro do grupo de doenças raras, tendo em vista que sua estimativa epidemiológica é de 6 a 7 em cada 100.000 nascimentos (SPONER; KORBEL; KUCERA, 2022).
Fisiopatologia da OI
Os ossos, junto com as cartilagens, ligamentos e tendões constituem o sistema esquelético, responsável por diversas funções vitais ao nosso corpo como: suporte, proteção, movimentação, homeostasia, hematopoese e armazenamento de gordura. O tecido ósseo contém uma matriz extracelular composta por água, fibras colágenas e sais minerais cristalizados. Conforme os cristais de sais são depositados na estrutura formada pelas fibras de colágeno, o tecido endurece através de um processo chamado de calcificação, iniciado a partir dos osteoblastos (células formadoras de ossos). Apesar da consistência sólida do osso depender da presença de sais minerais cristalizados em sua composição, a flexibilidade e resistência a tração, ou seja, resistência ao estiramento depende das fibras de colágeno, que, na OI, tem quantidade ou estrutura alteradas (TORTORA; DERRICKSON, 2016).
Além disso, destaca-se a presença de quatro tipos celulares no tecido ósseo, cada um com funções bem delimitadas. As células osteogênicas, também conhecidas como osteoprogenitoras são células tronco mesenquimatosas não especializadas e são as únicas células do tecido ósseo que sofrem divisão celular e originam os osteoblastos. Os osteoblastos são as células que sintetizam a parte orgânica da matriz óssea (colágeno tipo I, proteoglicanos e glicoproteínas) e participam da mineralização da matriz. As células responsáveis pela troca de nutrientes e resíduos com o sangue são chamadas de osteócitos e são essenciais para a manutenção da matriz óssea. Por último, as células denominadas osteoclastos são responsáveis pela reabsorção e remodelamento ósseo, sendo caracterizados como células gigantes e móveis que pertencem ao sistema fagocitário mononuclear (TORTORA; DERRICKSON, 2016; ROSS; PAWLINA, 2016).
O desenvolvimento ósseo é classificado de duas formas, ossificação intramembransa e endocondral, que se distinguem pelo modelo de cartilagem que formará o tecido ósseo. A ossificação intramembranosa é iniciada pela condensação de células mesenquimais a partir da oitava semana de gestação e que, posteriormente se diferem em osteoblastos – células secretoras principalmente de colágeno tipo I. A medida que o processo tem continuidade e a mineralização acontece, osteoblastos são diferenciados em osteócitos, formando ossos maduros, compactos e lamelares. Ossos planos do crânio e da face, mandíbula e clavícula desenvolvem-se através da ossificação intramembranosa, sendo essa classificação relevante também para o crescimento dos ossos curtos e aumento da espessura de ossos longos (ROSS; PAWLINA, 2016).
A ossificação endocondral também tem seu início com as células mesenquimatosas, mas, através da influência de diversos fatores de crescimento, essas células são diferenciadas em condroblastos, que expressam colágeno tipo II e produzem matriz cartilaginosa. Essa forma de ossificação começa no segundo trimestre de vida intrauterina e continua até a vida adulta quando o indivíduo atinge seu crescimento máximo. Ossos dos membros e das partes do esqueleto axial desenvolvem-se por ossificação endocondral (ROSS; PAWLINA, 2016).
A estrutura óssea está em constante remodelamento desde o nascimento até aproximadamente os 40 anos de vida. Esse processo está relacionado com a formação e reabsorção óssea, e varia de um indivíduo para outro de acordo com fatores genéticos e pico de massa óssea, além do estilo de vida, prática de atividades físicas, aporte nutricional, presença de doenças crônicas, endócrinas ou ósseas e uso de medicamentos. Dessa forma, tais fatores devem ser levados em consideração durante avaliação e suspeição de doenças com manifestações ósseas como a OI (MARQUES et al., 2016)
O colágeno tipo I é o principal componente proteico da matriz extracelular dos ossos, tendões e pele, sendo secretado principalmente por osteoblastos, fibroblastos e tenócitos (MARINI et al., 2017). Uma molécula de colágeno tipo I consiste em três cadeias polipeptídicas (duas cadeias alfa 1 e uma cadeia alfa 2) que formam uma estrutura helicoidal, sendo necessário um resíduo de glicina na terceira posição para que as três cadeias se entrelacem. Anormalidades pontuais que afetam um resíduo de glicina em COL1A1 ou COL1A2 são típicas da OI, e, nesses casos, as células que abrigam as mutações produzem colágeno anormal e normal. Dependendo de qual das cadeias será afetada, da posição da tripla hélice e qual aminoácido será substituído por glicina, o fenótipo da doença poderá variar entre leve a letal (ROSSI; LEE; MAROM, 2019).
As mutações em COL1A1 ou COL1A2 podem resultar em defeitos quantitativos que são caracterizados pela quantidade (síntese) reduzida de colágeno tipo I normal, ou em defeitos qualitativos em que há produção de colágeno com estrutura alterada (MARINI et al., 2017). De maneira geral, os defeitos quantitativos estão relacionados a OI do tipo mais leve, enquanto os defeitos qualitativos estão associados aos tipos considerados moderados e progressivos (HOLTZ et al., 2023).
Além disso, mutações estruturais podem ter como consequência a retenção prolongada de cadeias do colágeno tipo I desdobradas no retículo endoplasmático (RE), expondo-as às enzimas modificadoras pós-traducionais por tempo prolongado, resultando em matriz extracelular aberrante – característica principal subjacente a fragilidade óssea na OI. Em outros casos, o colágeno normal é parcialmente retido no RE estimulando a autofagia, ativação de apoptose e diferenciação osteoblástica prejudicada devido ao estresse ocasionado na organela. Por conseguinte, há aumento da disfunção da matriz óssea e redução da síntese de colágeno com consequente diminuição da massa óssea, caracterizando a fisiopatologia da osteogênese imperfeita (MARINI et al., 2017).
Classificação da OI
Tendo em vista a variabilidade de manifestações que a OI pode apresentar, no final da década de 1970, deu-se o início de estudos clínicos com intuito de classificar a doença (SILLENCE et al., 1979 apud MARINI et al., 2017). Assim, no ano de 1979, foi publicada a nosologia e classificação da OI em um estudo epidemiológico e genético intitulado “Heterogeneidade genética na osteogênese imperfeita”. A partir de então a OI passou a ser classificada por suas características clinicas, radiológicas e padrão de herança em quatro grupos, tipo I, II, III e IV (VAN DIJK; SILLENCE, 2014).
Dessa forma, há mais de duas décadas utiliza-se o padrão proposto de tipos I a IV, considerados a classificação da OI clássica. Essa forma de classificação tem como base as mutações nos genes COL1A1 e COL1A2 e suas consequentes características clínicas e radiográficas, sendo a mais aceita até os dias atuais. A classificação apresenta-se de tal maneira: tipo I, considerado a forma leve das apresentações da OI; tipo II, a forma perinatal letal; tipo III, variedade deformante progressiva e tipo IV, considerada a forma moderada da OI (MARINI et al., 2017).
Todavia, conforme se deram os avanços em estudo genéticos, em 2004 foi publicada uma expansão da classificação de Sillence, adicionando outros tipos de OI, abragendo defeitos genéticos que não envolvem os genes COL1A1 e COL1A2 do colagéno tipo I. Essas outras alterações genéticas são caracterizadas por serem de herança autossômica dominante presumida do gene IFITM5 (tipo V) e herança autossômica recessiva envolvendo o gene SERPINF1 e CRTAP (tipo VI e VII, respectivamente). Essa nova classificação ficou conhecida como a “classificação de Sillence expandida” e reconhece 7 tipos de OI (I a VII), ou seja, abrange a classificação clássica de Sillence com adição dos três outros tipos (Tabela 1) (VAN DIJK; SILLENCE, 2014).
Tabela 1 – Classificação expandida de Sillence da OI.
TIPO GRAVIDADE CLÍNICA MANIFESTAÇÕES TÍPICAS MUTAÇÕES ASSOCIADAS* I OI leve não deformante Altura normal ou leve baixa estatura, esclera azulada, sem dentinogênese imperfeita aparente Códon de parada prematuro em COL1A1 II Perinatal letal Multiplas fraturas de costelas e ossos longos ao nascimento, deformidades pronunciadas, ossos longos largos, esclera escura Substituições de glicina em baixa densidade em COL1A1 ou COL1A2 III Deformante progressivo Estatura gravamente baixa rostro triangular, escoliose grave, esclera acizentada, dentinogênese imperfeita Substituições de glicina em COL1A1 ou COL1A2 IV Moderadamente deformado Estatura moderadamente baixa, escoliose (leve ou moderada), esclera acizentada ou branca, dentinogênese imperfeita Substituições de glicina em COL1A1 ou COL1A2 V Moderadamente deformado Baixa estatura (leve ou moderada), luxação da cabeça do rádio, membrana interóssea mineralizada, calo hiperplásico, esclera branca, sem dentinogênese imperfeita IFITM5 VI Moderada a severamente deformante Estatura moderadamente baixa, escoliose, acúmulo de osteóide no tecido ósseo, lamelação óssea em padrão escama de peixe, esclera branca, sem dentinogênese imperfeita SERPINF1 VII Moderadamente deformante Baixa estatura leve, fêmures e úmeros curtos, coxa vara, esclera branca e sem dentinogê nese imperfeita CRTAP
*Pode não ser detectado em determinados pacientes.
Fonte: Adaptado de Rossi; Lee; Marom (2019); Alves (2019).
Os estudos e revisões tiveram continuidade e atualmente são descritos diversos tipos de OI, que, embora não apresentem defeitos nos genes COL1A1 e COL1A2, caracterizam-se por fragilidade óssea. Dessa forma há uma gama de classificações que relacionam as descobertas genéticas e os envolvimentos de loci gênicos às manifestações clínicas e resultados de exames de imagem (VAN DIJK; SILLENCE, 2014). Os tipos de OI bem como a relação entre as mutações e suas manifestações estão descritos na Tabela 2.
Tabela 2 – Classificação da OI de acordo com as características genéticas e clínicas.
TIPO HERANÇA GENE PROTEÍNA DEFEITO FENÓTIPO I AD COL1A1/COL1A2 Alfa1 do colágeno tipo I Quantidade do colágeno Moderado, não deformante II AD COL1A1/COL1A2 Alfa1/alfa2 do colágeno tipo I Estrutura do colágeno Perinatal letal III AD COL1A1/COL1A2 Alfa1/alfa2 do colágeno tipo I Estrutura do colágeno Deformante progressivo IV AD COL1A1/COL1A2 Alfa1/alfa2 do colágeno tipo I Estrutura do colágeno Moderadamente deformante V AD IFITM5 BRIL Mineralização da matriz óssea Moderado, histologia óssea típica, deslocamento da cabeça do rádio, calo hiperplásico, ossificação da membrana interóssea VI AR SERPINF1 PEDF Mineralização da matriz óssea Moderado a grave, histologia óssea típica em “escama de peixe”, acúmulo de osteoide VII AR CRTAP CRTAP Hidroxilação prolyl 3 Grave a letal. Coxa vara, úmero e fêmures curtos VIII AR LEPRE1 P3H1 Hidroxilação prolyl 3 Grave a letal IX AR PPIB CyPB Hidroxilação prolyl 3 Moderado a letal X AR SERPINH1 HSP47 Chaperoning do colágeno Grave XI AR FKBP10 FKBP65 Hidroxilação do telopeptídeo Deformante progressiva com contraturas congênitas (síndrome de Bruck. Pode estar associada a epidermólise bolhosa XII AR SP7 SPT/osterix Desenvolvimento do osteoblasto Moderado XIII AR BMP1 BMP1/mTLD Processamento do colágeno Grave, aumento da massa óssea XIV AR TEMEM38B TRIC-B Defeito no canal de cátion Moderado a grave XV AR WNT1 WNT1 – Variável XV AR WNT1 WNT1 – Início precoce de osteoporose
Legenda: Padrão autossômico dominante (AD), padrão autossômico recessivo (AR).
Fonte: Adaptado de Alves (2019).
Levando em consideração os fatores demonstrados acima, houve uma nova proposta de classificação, agora denominada metabólica funcional que objetiva uma maior utilidade e retenção da numeração do tipo genético. De acordo com essa classificação, os genes cujos produtos atuam na mesma via e tem mecanismos semelhantes ou compartilhados seriam organizados em cinco grupos funcionais: defeitos primários na estrutura ou processamento do colágeno (grupo A); defeitos na modificação do colágeno (grupo B); defeitos de dobramento e reticulação do colágeno (grupo C); defeitos de ossificação ou mineralização (grupo D); e defeitos no desenvolvimento de osteoblastos com insuficiência de colágeno (grupo E) (FORLINO; MARINI, 2016).
Para auxiliar a organização clínica, algumas das novas propostas genéticas foram mescladas com a proposta anterior de Sillence, por exemplo, o tipo II letal incluiu mutações letais de colágeno e algumas outras mutações em outros genes. Ainda assim, algumas classificações clínicas foram mantidas como antes esquematizadas, citando nesse caso os tipos V, VI e VII (FORLINO; MARINI, 2016). Entretanto, independetemente da etiologia molecular em que a nosologia se baseou, a classificação clínica/clássica de Sillence é ainda a mais comumente utilizada para determinar o tratamento e prognóstico dos pacientes, haja visto que a maior parte (85-90%) dos casos de OI se enquadram nos padrões dos subtipos I a IV devido às mutações nos genes COL1A1 ou COL1A2 (ROSSI; LEE; MAROM, 2019).
Manifestações Clínicas da OI
Como já descrito, a OI é considerada uma doença sistêmica do tecido conjuntivo, que se apresenta como uma displasia esquelética caracterizada primariamente por fragilidade e baixa densidade óssea com elevada incidência de fraturas. As fraturas podem envolver localizações atípicas se comparado a população geral, cita-se como exemplo as fraturas vertebrais que ocorrem em cerca de 70% dos pacientes com OI (ROSSI; LEE; MAROM, 2019). Ressalta-se ainda que, crianças e adolescentes com osteogênese imperfeita tem 11 vezes mais fraturas do que a população em geral, da mesma idade e sexo, devido aos efeitos das variantes genéticas desse distúrbio (FOLKESTAD et al., 2016).
Todavia, por se tratar de um distúrbio do colágeno, uma proteína presente em abundância em diversos tecidos corporais a doença apresenta também manifestações extraesqueléticas. Essas manifestações caracterizam-se pela presença de esclera azulada, dentinogênese imperfeita, graus de perda auditiva, comprometimento pulmonar, disfunção de válvulas cardíacas, fraqueza muscular e frouxidão ligamentar (MARINI et al., 2017).
Além disso, as anormalidades anatômicas e funcionais da parede torácica estão relacionadas intrinsicamente a limitação da função respiratória em pacientes com OI, devido à restrição diafragmática por alterações biomecânicas do tórax. Ainda, alguns estudos apontam para um possível acometimento intrínseco do pulmão na OI, tendo em vista a importância do colágeno tipo I no desenvolvimento do parênquima pulmonar. Nesse viés, o comprometimento da função respiratória na OI tem sido apontado como um grave fator para morbimortalidade nesses indivíduos (CARVALHO et al., 2023). Tais características clínicas, somadas as novas descobertas genéticas, são as utilizadas atualmente para classificar os pacientes com OI.
Entretanto, reafirma-se que o espectro das manifestações clínicas varia desde indivíduos assintomáticos ou com leve predisposição a fraturas, deformidades ósseas moderadas a graves progressivas, até a letalidade perinatal (MEI; ZHANG; ZHANG, 2022).
Pacientes com OI tipo I geralmente apresentam a forma leve da doença, sem deformidades ósseas. Esse tipo é subdividido em A e B, sendo que A é caracterizado por ausência da dentinogênese imperfeita e B refere-se a presença dessa manifestação. Ainda, o tipo I apresenta escleras azuladas e pode haver uma forma leve de escoliose como consequência das fraturas vertebrais, que ocorrem geralmente antes da puberdade. Normalmente não há comprometimento auditivo nesses pacientes, mas há afinamento da pele e hipermobilidade de articulações. Por se tratar de um caso considerado leve dentre as apresentações da OI, não há alteração na sua expectativa de vida (ALTALIB et al., 2021).
Em relação ao tipo II, infere-se que as características apresentadas são extremamente graves, tendo como consequência a letalidade perinatal. A principal apresentação dos fetos acometidos são deformidades em curvatura ou angulação dos ossos longos e deficiência de ossificação dos ossos faciais e do crânio (VAN DIJK; SILLENCE, 2014). Além disso, com o constante crescimento intrauterino, há fraturas progressivas de costelas que, por conseguinte, ocasionam insuficiência respiratória, a principal causa da letalidade nos casos de OI tipo II. Outros sinais comuns a essa classe são a esclera azulada e múltiplas fraturas e deformidades de extremidades (ALTALIB et al., 2021).
A OI do tipo III é denominada como progressivamente deformante, isso se deve ao fato de que, indivíduos assim classificados apresentam elevada fragilidade óssea com progressiva deformidade, osteopenia generalizada e fraturas que podem ser observadas em estudos de imagem logo ao nascimento. Ainda, os pacientes podem ter manifestações de esclera azulada ou acizentada, dentinogênese imperfeita e, perda auditiva, que pode não estar presente na infância, mas tende a ocorrer na vida adulta. Em consequência da progressiva deformidade e fragilidade óssea, indivíduos com OI tipo III apresentam baixa estatura e escoliose, entretanto, sobrevivem até a vida adulta (PALOMO; VILAÇA; LAZARETTI-CASTRO, 2017).
O tipo IV tem gravidade moderada e caracteriza-se por leve ou moderada fragilidade óssea, podendo apresentar ou não baixa estatura e escoliose. As escleras são normais ou acinzentadas, e esses pacientes apresentam perda auditiva. O tipo V apresenta baixa estatura e, devido a limitação do movimento no antebraço pode apresentar deslocamento da cabeça do rádio. Além disso, possuem membrana interóssea mineralizada e calo hiperplásico. O tipo VI tem seus critérios baseados na histologia: acúmulo de osteoide (matriz óssea que ainda não foi calcificada) e padrão de lamelação óssea deformante, a clínica envolve a presença de esclera branca e moderada estatura. Por último, o tipo VII apresenta moderada gravidade e é definido por manifestar esclera branca, pouca redução de estatura, úmero e fêmur encurtados além de coxa vara (BRASIL, 2022).
Além dessas manifestações clínicas, também há relatos de dores agudas e crônicas pelos pacientes, sendo a dor crônica mais prevalente e geralmente associada a uma diminuição na qualidade de vida dos pacientes (PROVENZANO; ÅSTRÖM; LÖWING, 2022). A dor crônica não decorrente de fraturas ou lesões é frequentemente relatada por crianças e adolescentes, sendo responsável pela mudança no padrão de atividades diárias desses pacientes, que acabam por se tornar menos ativos devido à intensidade do quadro doloroso. (CORTÉS; PASTOR; DOLZ, 2022).
Métodos Diagnósticos da OI
A suspeição da OI é decorrente de uma avaliação clínica, que requer semelhanças com a história e apresentações clínicas da doença. Por se tratar de um distúrbio heterogêneo com diversas manifestações, o diagnóstico não pode ser essencialmente clínico. Sendo assim, é importante investigar a determinação da síntese reduzida ou anormal de moléculas de colágeno do tipo I, por meio de cultura de fibroblastos ou através da identificação de mutações nos genes COL1A1 e COL1A2 que codificam as cadeias do colágeno tipo I, para então efetuar a identificação e confirmação da OI (MORABITO et al., 2022).
Além do exame clínico e testes genéticos, avaliações laboratoriais podem auxiliar no diagnóstico e condutas terapêuticas. Dentre essas avaliações, dosagens de cálcio, fósforo, fosfatase alcalina e paratormônio são alguns dos exames que podem ser solicitados para avaliação sérica do metabolismo do cálcio. Também faz parte dos métodos de diagnósticos para OI a radiografia, sendo indicada para identificação de fraturas, calos e deformidades ósseas. O exame radiográfico deve ser solicitado em incidências anteroposterior (AP) e perfil dos ossos simples longos e, também panorâmica da coluna em AP e perfil. Ressalta-se que a união dos achados clínicos e radiológicos são a base para o diagnóstico da osteogênese imperfeita (BRASIL, 2022).
Mesmo que o diagnóstico da OI dependa dos fatores e exames demonstrados acima, sabe-se que alguns pacientes acometidos pela osteogênese imperfeita podem ter seu diagnóstico ainda no período pré-natal. A triagem para a osteogênese imperfeita limita-se ao exame de ultrassonografia no pré-natal, podendo detectar a forma perinatal letal (tipo II) entre as 14 e 16 semanas de gestação. A identificação do tipo III ocorre por volta de 18 semanas, quando há um declínio no percentil do crescimento longitudinal. Por vezes, o sequenciamento genético de amostras de vilosidades coriônicas é utilizado para confirmar a presença de OI em indivíduos com histórico familiar do distúrbio (MARINI et al., 2017).
Todavia, salienta-se que o diagnóstico da osteogênese não é simples, podendo alguns distúrbios esqueléticos primários serem confundidos com OI. Nesse caso, é necessária a exclusão da osteoporose idiopática ou juvenil, por se apresentarem como diagnósticos diferenciais da OI. Além disso, crianças com graus de fragilidade óssea sem manifestações características de OI também devem receber uma avaliação cuidadosa. Por último, pode se destacar o abuso infantil como uma das mais notáveis causas de fratura, sendo a maior incidência no primeiro ano de vida com sinais de fraturas em costelas e fêmur principalmente, a OI está presente em 2 a 5% dos casos desse contexto. (PALOMO; VILAÇA; LAZARETTI-CASTRO, 2017).
Tratamento da OI
Atualmente a gestão terapêutica da OI visa reduzir a incidência de fraturas, melhorar a função motora e a qualidade de vida dos pacientes através da inclusão social, haja visto que ainda não há cura para a OI. Para que isso ocorra de maneira efetiva é preciso que a abordagem seja multidisciplinar, envolvendo uma variedade de profissionais de saúde como médicos, fisioterapeutas, psicólogos, entre outros (ROSSI; LEE; MAROM, 2019).
A abordagem terapêutica para pacientes com OI envolve algumas variantes como a idade, o estado funcional do paciente e a gravidade da doença. Por exemplo, indivíduos com OI tipo I (leve) podem viver com pouca ou nenhuma restrição e geralmente a terapia ortopédica é suficiente para o tratamento das fraturas. No entanto, pacientes com tipos moderados a graves necessitam de maiores restrições, reabilitação e intervenções (PALOMO; VILAÇA; LAZARETTI-CASTRO, 2017).
A resistência óssea é dependente de algumas propriedades dos ossos, como por exemplo a qualidade do material ósseo, a quantidade de massa óssea e a forma como está distribuída. Na OI, a matriz óssea apresenta-se de forma desorganizada e hipermineralizada devido a presença de um maior número de osteoblastos, responsáveis pela síntese da matriz orgânica dos ossos e osteoclastos responsáveis pelo remodelamento e reabsorção óssea. Tal característica tem como resultado a fragilidade óssea, responsável pelas características clínicas da OI. Atualmente não há nenhuma terapia que atue alterando diretamente essa forma de organização óssea da OI, todavia, terapias antirreabsortivas e anabólicas apresentam resultados de melhora do volume ósseo (MARINI et al., 2017).
Em relação as terapias antirreabsortivas, cita-se os bifosfonatos (alendronato, risedronato, pamidronato e ácido zoledrônico) como base dos cuidados farmacológicos em pacientes pediátricos com OI. Esses fármacos melhoram a massa e arquitetura óssea, tendo como principal resultado a redução dos riscos de fraturas, pois seu mecanismo de ação baseia-se na inibição da atividade e indução de apoptose dos osteoclastos, além de auxiliar na redução do quadro doloroso dos pacientes. Esses medicamentos podem ser administrados via intravenosa e oral. Os fármacos de administração intravenosa parecem possuir efeitos superiores na melhora da densidade óssea e redução da taxa de fraturas, além de auxiliar na redução da dor crônica e ocasionar menores efeitos colaterais (ROSSI; LEE; MAROM, 2019).
No entanto, a utilização de bifosfonatos é responsável por alguns efeitos adversos como desconforto e irritação gastrointestinal quando administrados por via oral. Com relação a via intravenosa os principais efeitos adversos são febre, dores musculares e vômitos relacionados a primeira infusão, ainda, pode ocorrer uma diminuição do cálcio sérico transitória imediatamente após a infusão. Todavia, a terapia com bisfosfonatos é bem tolerada e seus benefícios já são bem estabelecidos no tratamento clínico da OI (PALOMO; VILAÇA; LAZARETTI-CASTRO, 2017).
Devido ao potencial de ação anabólico do hormônio do crescimento – Growth Hormone (GH) no osso, esse tem sido proposto como uma possibilidade terapêutica para a OI objetivando principalmente minimizar a deficiência de crescimento apresentada pela maioria dos indivíduos. A utilização do hormônio GH apresenta sinergia quando combinado com bifosfonatos, aumentando a velocidade de crescimento, porém, não apresentou o mesmo efeito na redução de riscos de fraturas. Atualmente, essa abordagem não é padronizada no tratamento clínico, necessitando de maiores investigações. Quanto ao uso da terapia com anabolizantes em OI, essa ainda é aprovada apenas para o uso em adultos com osteoporose, visando o aumento da formação óssea (ROSSI; LEE; MAROM, 2019).
Outros agentes terapêuticos estão sendo estudados para utilização na OI, como o denosumabe, um anticorpo monoclonal humano que tem como mecanismo de ação o bloqueio do RANKL, uma das citocinas essenciais na via da osteoclastogênese. O resultado é basicamente o mesmo dos bifosfonatos, promovendo a inibição de osteoclastos, com consequente supressão da reabsorção óssea. Por outro lado, a terapia celular com direcionamento genético, através da supressão de transcrições prejudiciais ou adição de genes ou transplante de células pode exercer grande relevância na gestão da OI, todavia, ambos os tratamentos necessitam de maiores pesquisas para obtenção de melhores resultados (FRANZONE et al., 2019; SCHINDELER et al., 2022).
De acordo com o Protocolo Clínico e Diretrizes Terapêuticas da OI no Brasil, o alendronato oral e pamidronato dissódico de uso intravenoso (bisfosfonatos) são as medicações aprovadas e protocoladas para pacientes com OI que apresentam deformidades de ossos longos, fraturas por compressão vertebral e que tenham sofrido mais de duas fraturas por ano, além da suplementação com cálcio e vitamina D. Espera-se como benefícios através desse esquema terapêutico a redução da incidência de fraturas, melhora da dor crônica, redução da incapacidade física, melhora da mobilidade e crescimento, e ainda a prevenção e tratamento da hipocalcemia. O tempo de tratamento recomendado é por 2 anos após o paciente não apresentar mais fraturas (BRASIL, 2022; ALVES, 2019).
A gestão ortopédica da OI visa atingir marcos de desenvolvimento o mais próximo do considerado saudável e padrão, aumentar a função óssea e reduzir a incidência de fraturas e de suas consequências. Para isso, a atividade física tem se mostrado como uma peça principal, melhorando a capacidade funcional e força muscular dos pacientes. Nesse sentido, a fisioterapia apresenta-se como aliada ao manejo ortopédico. O plano terapêutico desenvolvido por fisioterapeutas deve ser individualizado conforme a apresentação do paciente. Desde a infância até a vida adulta, objetiva-se atingir marcos de desenvolvimento e maximizar a mobilidade dos pacientes, tendo como resultado final a independência, qualidade de vida e inclusão social (FRANZONE et al., 2019).
Em relação aos procedimentos cirúrgicos ortopédicos, o realinhamento com osteotomia com haste intramedular demonstra-se como a melhor cirurgia dos membros inferiores em pacientes com OI, tratando-se da inserção de uma haste metálica na cavidade medular do osso com o intuito de fornecer maior força e consequente alinhamento. Nos membros superiores o procedimento de escolha geralmente é o roding de úmero com proteção do nervo radial, no qual podem ser utilizadas hastes para permitir maior alinhamento e funcionalidade dos membros (MARINI et al., 2017).
A psicologia é outra área que ocupa um espaço de grande relevância dentro da multidisciplinariedade do tratamento e acompanhamento dos pacientes com OI. Conforme as crianças se desenvolvem e crescem, começam a se deparar com situações que demandam suporte emocional, físico e social. A avaliação psicológica dos pacientes com OI e de suas famílias pode e deve ser realizada pelo psicólogo, que é um profissional capacitado para oferecer o suporte adequado para esses indivíduos e seus familiares. Consultas psicológicas auxiliam na delimitação de estratégias para alcançar a estabilidade emocional, além de possibilitar para a equipe de profissionais envolvidos no caso uma visão mais psíquica das limitações desses pacientes (MARR; SEASMAN; BISHOP, 2017).
Por fim, no Brasil, preconiza-se de acordo com a faixa etária que crianças de até dois anos de idade, de dois a três anos, com mais de três anos e adultos tenham consultas médicas e avaliações multiprofissionais realizadas a cada dois meses, a cada três meses, a cada 4 meses e a cada 6 meses respectivamente. É importante ressaltar a necessidade da avaliação do tratamento medicamentoso em todas as consultas, levando em consideração exames laboratoriais e radiológicos, além de manter rotina com médicos especialistas conforme as manifestações extraesqueléticas dos pacientes com OI (BRASIL, 2022).
Considerações Finais
A osteogênese imperfeita (OI) é uma doença genética que se apresenta de forma rara, com uma complexa fisiopatologia e diversificadas manifestações clínicas e radiológicas. O desconhecimento a respeito da doença e suas formas clínicas atua como um fator atenuante para o atraso do diagnóstico e tratamento, acarretando em dificuldades no crescimento e desenvolvimento dos indivíduos que vivem com a OI.
Neste estudo foi descrito que mutações que codificam o colágeno do tipo I são as principais responsáveis pela OI, onde os pacientes acometidos podem apresentar manifestações esqueléticas e extraesqueléticas, podendo ser desde formas mais brandas até graves e letais. Assim, a OI pode ser classificada de acordo com suas alterações genéticas e manifestações clínicas. O diagnóstico normalmente é realizado pelo exame clínico, exames de imagem, laboratoriais e genéticos. Ademais, diferentes formas terapêuticas podem ser utilizadas de forma individualizada para cada paciente, podendo ser utilizado fármacos que influenciam no metabolismo ósseo, suplementação com cálcio e vitamina D, controle da dor, reposição hormonal e até mesmo o uso de imunoterapia, essa última ainda necessita de mais estudos, no entanto, algumas pesquisas tem apresentado resultados promissores (Figura 1).
Dessa maneira, o diagnóstico precoce da OI associado a instituição do tratamento adequado e efetivo, contribuem com o melhor prognóstico da doença e qualidade de vida dos indivíduos acometidos pela doença. Esta revisão destacou os avanços sobre a OI, sua fisiopatologia, manifestações clínicas, classificações, diagnóstico e tratamentos, podendo contribuir para ciência e para os profissionais de saúde, bem como incentivar mais pesquisas sobre OI.
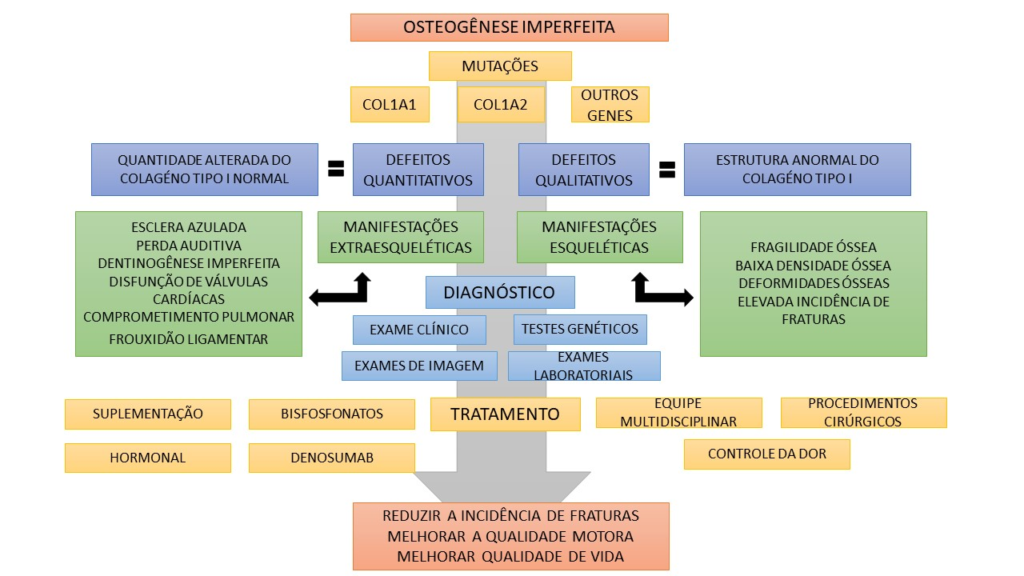
Figura 1 – Fluxograma descritivo sobre a OI. A Osteogênese imperfeita é causada por mutações nos genes COL1A1, COL1A2 e outros genes relacionados com a expressão do colágeno do tipo I. Essas mutações podem ocasionais em defeitos quantitativos, onde há uma menor síntese do colágeno do tipo I, e defeitos qualitativos, quando ocorre a síntese do colágeno do tipo com estrutura anormal. Além disso, indivíduos com OI podem apresentar manifestações esqueléticas e extraesqueléticas. O diagnóstico dessa doença pode ser realizado por meio do exame clínico, exames de imagem e laboratoriais, além de testes genéticos para avaliar as mutações. O tratamento pode ser realizado utilizando bisfosfonatos, suplementação, procedimentos cirúrgicos, fármacos para controle da dor, reposição hormonal e também o uso de Denosumab (anticorpo monoclonal, ainda não utilizado rotineiramente). A terapia tem como objetivo reduzir a incidência de fraturas, melhorar a qualidade motora e de vida dos pacientes. Fonte: O autor (2023).
ALTALIB, Abdulraheem. et al. Osteogenesis Imperfecta and Child Abuse From a Forensic Point of View. Cureus, [S.L.], v. 1, n. 13, p. 1-11, 19 jan. 2021. Cureus, Inc.. http://dx.doi.org/10.7759/cureus.12790. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7891677/. Acesso em: 17 set. 2022
ALVES, Crésio de Aragão Dantas. ENDOCRINOLOGIA PEDIÁTRICA. Barueri: Manole, 2019. 834 p. Disponível em: https://integrada.minhabiblioteca.com.br/reader/books/9788520458020/pageid/3. Acesso em: 26 set. 2022.
BARLOW, Sophie; DOVE, Lucy; JAGGI, Anju; KEEN, Richard; BUBBEAR, Judith. The prevalence of musculoskeletal pain and therapy needs in adults with Osteogenesis Imperfecta (OI) a cross-sectional analysis. Bmc Musculoskeletal Disorders, [S.L.], v. 23, n. 1, p. 1-7, 21 maio 2022. Springer Science and Business Media LLC. http://dx.doi.org/10.1186/s12891-022-05433-3. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9123157/. Acesso em: 30 mar. 2023.
BRASIL, Ministério da Saúde. Protocolo Clínico e Diretrizes Terapêuticas da Osteogênese Imperfeita. Brasilia: Conitec, 2022. 57 p. Disponível em: https://www.gov.br/conitec/pt-br/midias/consultas/relatorios/2022/20220527_relatorio_pcdt_osteogenese_imperfeita_cp_32.pdf. Acesso em: 17 set. 2022.
BRASIL, Ministério da Saúde. Doenças raras. 2022. Disponível em: https://www.gov.br/saude/pt-br/assuntos/saude-de-a-a-z/d/doencas-raras-1. Acesso em: 14 set. 2022.
CARVALHO, Patricia de Abreu Farias; REGIS, Taiane Sousa; FAIÇAL, Adriana Virgínia Barros; TERSE-RAMOS, Regina; ACOSTA, Angelina Xavier. Respiratory function of children and adolescents with osteogenesis imperfecta: respiratory muscle strength, forced vital capacity, and peak expiratory flow. Revista Paulista de Pediatria, [S.L.], v. 41, p. 1-5, 2023. FapUNIFESP (SciELO). http://dx.doi.org/10.1590/1984-0462/2023/41/2022092. Disponível em: https://www.scielo.br/j/rpp/a/wtJGvJ9rDsLgnxJNLncnXLc/?lang=en. Acesso em: 20 abr. 2023.
CORTÉS, Rubén Muñoz; PASTOR, José Francisco Soriano; DOLZ, Vicente Monsalve. Chronic pain in adults with osteogenesis imperfecta and its relationship to appraisal, coping, and quality of life: a cross-sectional study. Medicine, [S.L.], v. 101, n. 40, p. 1-11, 7 out. 2022. Ovid Technologies (Wolters Kluwer Health). http://dx.doi.org/10.1097/md.0000000000030256. Disponível em: https://pubmed.ncbi.nlm.nih.gov/36221335. Acesso em:17 set. 2022.
FOLKESTAD, Lars. et al. Fracture Rates and Fracture Sites in Patients With Osteogenesis Imperfecta: a nationwide register-based cohort study. Journal Of Bone And Mineral Research, [S.L.], v. 32, n. 1, p. 125-134, 29 ago. 2016. Wiley. http://dx.doi.org/10.1002/jbmr.2920. Disponível em: https://asbmr.onlinelibrary.wiley.com/doi/10.1002/jbmr.2920. Acesso em: 17 set. 2022.
FORLINO, Antonella; MARINI, Joan C. Osteogenesis imperfecta. The Lancet, [S.L.], v. 387, n. 10028, p. 1657-1671, abr. 2016. Elsevier BV. http://dx.doi.org/10.1016/s0140-6736(15)00728-x. Disponível em: https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(15)00728-X/fulltext. Acesso em: 17 set. 2022.
FRANZONE, Jeanne M. et al. Osteogenesis Imperfecta. Orthopedic Clinics Of North America, [S.L.], v. 50, n. 2, p. 193-209, abr. 2019. Elsevier BV. http://dx.doi.org/10.1016/j.ocl.2018.10.003. Disponível em: https://www.sciencedirect.com/science/article/abs/pii/S0030589818301779?via%3Dihub. Acesso em: 18 set. 2022.
GARIBALDI, Nadia. et al. Dissecting the phenotypic variability of osteogenesis imperfecta. Disease Models & Mechanisms, [S.L.], v. 15, n. 5, p. 1-15, 1 maio 2022. The Company of Biologists. http://dx.doi.org/10.1242/dmm.049398. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9150118/. Acesso em: 14 set. 2022.
HALD, J.D. et al. Osteogenesis imperfecta and the teeth, eyes, and ears—a study of non-skeletal phenotypes in adults. Osteoporosis International, [S.L.], v. 29, n. 12, p. 2781-2789, 24 ago. 2018. Springer Science and Business Media LLC. http://dx.doi.org/10.1007/s00198-018-4663-x. Disponível em: https://pubmed.ncbi.nlm.nih.gov/30143849/. Acesso em: 14 set. 2022.
HOLTZ, A.P. et al. Genetic analysis of osteogenesis imperfecta in a large Brazilian cohort. Bone, [S.L.], v. 169, p. 116683, abr. 2023. Elsevier BV. http://dx.doi.org/10.1016/j.bone.2023.116683. Disponível em: https://www.sciencedirect.com/science/article/abs/pii/S8756328223000157?via%3Dihub. Acesso em: 20 abr. 2023.
MARINI, Joan C. et al. Osteogenesis imperfecta. Nature Reviews Disease Primers, [S.L.], v. 3, n. 1, p. 1-19, 18 ago. 2017. Springer Science and Business Media LLC. http://dx.doi.org/10.1038/nrdp.2017.52. Disponível em: https://www.nature.com/articles/nrdp201752. Acesso em: 20 ago. 2022.
MARQUES, Marcia Alessandra Arantes; LIMA, Daniely Alves; ANDREOTTI, Carlos Eduardo; GASPAROTTO JUNIOR, Arquimedes; LOURENÇO, Emerson Luiz Botelho. CARACTERIZAÇÃO DAS PLANTAS MEDICINAIS E MEDICAMENTOS FITOTERÁPICOS PARA TRATAMENTO DA OSTEOPOROSE UTILIZADOS NO BRASIL. Arquivos de Ciências da Saúde da Unipar, [S.L.], v. 20, n. 3, p. 183-188, 30 mar. 2016. Universidade Paranaense. http://dx.doi.org/10.25110/arqsaude.v20i3.2016.5870. Disponível em: https://ojs.revistasunipar.com.br/index.php/saude/article/view/5870/3382. Acesso em: 20 abr. 2023.
MARR, Caroline; SEASMAN, Alison; BISHOP, Nick. Managing the patient with osteogenesis imperfecta: a multidisciplinary approach. Journal Of Multidisciplinary Healthcare, [S.L.], v. 10, p. 145-155, abr. 2017. Informa UK Limited. http://dx.doi.org/10.2147/jmdh.s113483. Disponível em: https://pubmed.ncbi.nlm.nih.gov/28435282/. Acesso em: 25 ago. 2022.
MEI, Yazhao; ZHANG, Hao; ZHANG, Zhenlin. Comparing Clinical and Genetic Characteristics of De Novo and Inherited COL1A1/COL1A2 Variants in a Large Chinese Cohort of Osteogenesis Imperfecta. Frontiers In Endocrinology, [S.L.], v. 13, p. 1-10, 14 jul. 2022. Frontiers Media SA. http://dx.doi.org/10.3389/fendo.2022.935905. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9329653/. Acesso em: 25 ago. 2022.
MORABITO, Letteria Anna. et al. Osteogenesis Imperfecta/Ehlers–Danlos Overlap Syndrome and Neuroblastoma—Case Report and Review of Literature. Genes, [S.L.], v. 13, n. 4, p. 581, 25 mar. 2022. MDPI AG. http://dx.doi.org/10.3390/genes13040581. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9024599/. Acesso em: 25 ago. 2022.
PALOMO, Telma; VILAÇA, Tatiane; LAZARETTI-CASTRO, Marise. Osteogenesis imperfecta. Current Opinion In Endocrinology & Diabetes And Obesity, [S.L.], v. 24, n. 6, p. 381-388, dez. 2017. Ovid Technologies (Wolters Kluwer Health). http://dx.doi.org/10.1097/med.0000000000000367. Disponível em: https://pubmed.ncbi.nlm.nih.gov/28863000/. Acesso em: 25 ago. 2022.
PROVENZANO, Anna Hallin; ÅSTRÖM, Eva; LÖWING, Kristina. Exploring pain interference and self-perceived health status in children with osteogenesis imperfecta – a cross-sectional study. Bmc Musculoskeletal Disorders, [S.L.], v. 23, n. 1, p. 1-8, 21 set. 2022. Springer Science and Business Media LLC. http://dx.doi.org/10.1186/s12891-022-05825-5. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9490967/. Acesso em: 30 mar. 2023.
ROSS, Michael H.; PAWLINA, Wojciech. Histologia: texto e atlas: correlações com biologia celular e molecular. 7. ed. Rio de Janeiro: Editora Guanabara Koogan Ltda, 2016. 1000 p
ROSSI, Vittoria; LEE, Brendan; MAROM, Ronit. Osteogenesis imperfecta: advancements in genetics and treatment. Current Opinion In Pediatrics, [S.L.], v. 31, n. 6, p. 708-715, dez. 2019. Ovid Technologies (Wolters Kluwer Health). http://dx.doi.org/10.1097/mop.0000000000000813. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7017716/. Acesso em: 17 set. 2022.
SCHINDELER, Aaron. et al. Curative Cell and Gene Therapy for Osteogenesis Imperfecta. Journal Of Bone And Mineral Research, [S.L.], v. 37, n. 5, p. 826-836, 17 abr. 2022. Wiley. http://dx.doi.org/10.1002/jbmr.4549. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9324990/. Acesso em: 20 ago. 2022.
SPONER, Pavel; KORBEL, Martin; KUCERA, Tomas. Challenges of total knee arthroplasty in osteogenesis imperfecta: case report and literature review. Journal Of International Medical Research, [S.L.], v. 50, n. 5, p. 030006052210973, maio 2022. SAGE Publications. http://dx.doi.org/10.1177/03000605221097369. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9152202/. Acesso em: 20 ago. 2022.
TORTORA, Gerard J.; DERRICKSON, Bryan. PRINCIPIOS DE ANATOMIA E FISIOLOGIA. 14. ed. Rio de Janeiro: Guanabara Koogan Ltda, 2016. 1600 p.
VAN DIJK, F.s.; SILLENCE, D.O.. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. American Journal Of Medical Genetics Part A, [S.L.], v. 164, n. 6, p. 1470-1481, 8 abr. 2014. Wiley. http://dx.doi.org/10.1002/ajmg.a.36545. Disponível em: https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.36545. Acesso em: 17 set. 2022
Maria Eduarda Wilvert
UNIARP – Universidade Alto Vale do Rio do Peixe
mariawilvert@gmail.com
ORCID: https://orcid.org/0009-0006-7596-8587
Ariana Centa
UNIARP – Universidade Alto Vale do Rio do Peixe
ORCID: https://orcid.org/my-orcid?orcid=0000-0002-0419-141X
Maria Aparecida Marques Habermann
UNIARP – Universidade Alto Vale do Rio do Peixe
ORCID: https://orcid.org/0000-0002-0668-7849
João Paulo Assolini
UNIARP – Universidade Alto Vale do Rio do Peixe
ORCID: https://orcid.org/0000-0003-4190-8835