REGISTRO DOI: 10.5281/zenodo.10857443
Ana Beatriz Silva Sant’Ana¹;
Ana Luiza Forti Neves²;
Clarissa Torresan³.
RESUMO
Existe uma gama de genes que, quando mutados, originam síndromes que se enquadram dentro do transtorno do espectro autista (TEA). Dentre os principais, temos o gene MECP2, analisado em muitos estudos. Mutações nesse gene geram alterações em nível molecular que influenciam nos aspectos clínicos, repercutindo negativamente nos processos cognitivos e comportamentais. O objetivo dessa pesquisa foi apresentar o gene MECP2 e mutações descritas relacionadas à Síndrome de RETT e ao TEA, a fim de contribuir para a compreensão abrangente das bases moleculares e comportamentais dessas condições, oferecendo informações relevantes para diagnóstico e intervenção. O estudo foi realizado através de uma revisão integrativa acerca do tema, revelando resultados confluentes que sustentam as alterações neurológicas e comportamentais da RTT em diferentes níveis de análise. Isso ressalta a complexidade e interconexão de diferentes fatores que contribuem para essa síndrome neuropsiquiátrica.
PALAVRAS-CHAVE: Transtorno do espectro autista; TEA; autismo; genética; gene
ABSTRACT
There is a range of genes that, when mutated, give rise to syndromes that fall within the autism spectrum disorder (ASD). Among the main genes studied is MECP2. Mutations in this gene lead to molecular-level changes that impact clinical aspects, negatively affecting cognitive and behavioral processes. The aim of this research was to present the MECP2 gene and described mutations related to Rett Syndrome and ASD, contributing to a comprehensive understanding of the molecular and behavioral foundations of these conditions, providing relevant information for diagnosis and intervention. The study was conducted through an integrative review on the topic, revealing convergent results supporting the neurological and behavioral alterations of RTT at different levels of analysis. This highlights the complexity and interconnection of various factors contributing to this neuropsychiatric syndrome.
KEYWORDS: Autism spectrum disorder;ASD, autism; genetics; gene
1 INTRODUÇÃO
O transtorno do espectro autista (TEA) é um transtorno do neurodesenvolvimento que se caracteriza por déficits na interação e na comunicação social, juntamente com a presença de padrões nos comportamentos, interesses ou atividades classicamente restritos e repetitivos. Dentro do TEA existe o transtorno autista (autismo), o transtorno de Asperger, o transtorno desintegrativo da infância, o transtorno de Rett e o transtorno global do desenvolvimento sem outra especificação (AMERICAN PSYCHIATRIC ASSOCIATION, 2013).
Em 2020, foi feito um levantamento pela Rede de Monitoramento de Deficiências no Desenvolvimento e Autismo (ADDM) entre crianças de 8 anos em 11 locais dos Estados Unidos, no qual 1 a cada 36 crianças foi identificada com TEA, acometendo todos os grupos étnicos, raciais e socioeconômicos. Além disso, foi observado que o TEA ocorre quase quatro vezes mais em meninos do que em meninas (MAENNER, 2021). O sexo masculino é um dos fatores etiológicos mais bem estabelecidos para o TEA, originando a noção do efeito protetor feminino, que necessita de uma maior carga etiológica para que o mesmo grau de acometimento masculino, seja manifestado. Por esse motivo é que as mulheres estão propensas a exibir um fenótipo mais grave (ZEIDAN, 2022).
Em 2021, foi realizada uma busca para estimar a prevalência do TEA, com base em 70 estimativas, sendo a última realizada em 2012, de amplitude global, sendo constatado que a prevalência do TEA, em média, é de 62/10000 crianças. Há uma variação entre regiões, sendo que, algumas delas apresentaram-se muito limitadas ou completamente ausentes, como a Europa oriental e África. As regiões geográficas que apresentaram uma maior estimativa foram o pacífico ocidental, mediterrâneo oriental e américa com 203,1, 86,5 e 82,3 casos de TEA a cada 10000 habitantes, respectivamente (ZEIDAN, 2022).
Os fatores de riscos ambientais no TEA incluem a idade avançada dos pais, assim como suas histórias psiquiátricas, condições de nutrição, situação socioeconômica e o método de fertilização in vitro. Além disso, existem fatores relacionados à gestação, como estresse pré-natal, infecções, febre sem tratamento, diabetes gestacional, síndromes metabólicas, exposição pré-natal à poluição do ar ou a teratogênicos (medicamentos, drogas psicoativas, produtos químicos e plástico), hipóxia fetal, pré-eclâmpsia, prematuridade e pós-maturidade, parto por cesariana, nascimento de gêmeos, anomalias congênitas, complicações pós-natais, amamentação abaixo do ideal, entre outros (STYLES, 2020).
Como critérios diagnósticos, os sintomas devem ser precoces durante o desenvolvimento, não podendo ser explicados por outra condição médica ou psiquiátrica, além de necessariamente prejudicar alguma área significativa da vida do indivíduo, seja ocupacional, social ou outras (AMERICAN PSYCHIATRIC ASSOCIATION, 2013). Informações clínicas detalhadas, exames, dados de imagem e investigações físicas são importantes para traduzir achados genéticos, importantes para a prática clínica (THAPAR, 2021).
Os estudos de genética molecular reforçam que o TEA é uma condição heterogênea, tanto clinicamente quanto em termos de fatores causais genéticos. A partir do sequenciamento do exoma, foram observadas variantes raras de novo e herdadas relacionadas ao risco de TEA. No sequenciamento do genoma em famílias, mais da metade dos irmãos afetados carregam diferentes variantes relacionadas ao TEA (THAPAR, 2021).
Os conhecimentos obtidos no maior estudo de sequenciamento de exoma de TEA, realizado até o momento, incluíram a análise de 11.986 casos (6.430 trios probando-pais e 5.556 casos com 8.809 controles). A análise dos dados levou à pressuposição de 102 genes de risco para o TEA. (THAPAR, 2021).
De acordo com a Simons Foundation Autism Research Initiative (SFARI Gene), um banco de dados destinado aos pesquisadores do autismo, até o momento existem 1152 genes envolvidos na suscetibilidade ao autismo. Entre esses, temos o gene MECP2, localizado no braço longo do cromossomo X (Xq28).
O gene MECP2 é composto por quatro exons que podem codificar dois transcritos: MECP2-E1 e MECP2-E2. Ambos possuem um domínio de ligação a metil-CpG, um interdomínio, um domínio de repressão transcricional (TRD) e um domínio C-terminal (TA, 2022).
O gene MECP2 é responsável por codificar a proteína 2 de ligação ao metil-CpG (MECP2), que possui atividade repressora transcricional dependente de metilação e também está implicado na regulação do splicing de RNA. É expressa nos neurônios pós-natais e aumenta com a idade e a neurogênese. Mutações no gene MECP2 estão associadas à Síndrome de Rett (RTT), descrita em 1966 por Andreas Rett (TA, 2022).
O aconselhamento genético é importante quando uma variante patogênica do MECP2 é reconhecida em uma família pois, nesses casos, podem ser realizados testes heterozigotos para parentes do sexo feminino em risco, testes pré-natais para gestações de alto risco e testes genéticos pré-implantação. Além disso, em casos de mãe heterozigota conhecida, em que a prole apresenta 50% de risco de herdar a variante MECP2. Ademais, para casais que têm um filho com a mutação MECP2 é adequado oferecer diagnóstico pré-natal devido à possibilidade de mosaicismo da linha germinativa parental, independentemente de a variante patogênica MECP2 ter sido detectada em um dos pais (KAUR; CHRISTODOULOU, 2001).
Apenas a presença de uma mutação MECP2 não é suficiente para o diagnóstico de RTT, sendo necessárias características clínicas típicas ou atípicas, já que essa mutação pode causar outras condições neurológicas (NEUL, 2010).
No mundo, a prevalência dessa alteração genética é de 1:10.000-1:23.000 nascimentos do sexo feminino. As mulheres podem ter um espectro de fenótipos variando desde dificuldades de aprendizagem leves, devido ao padrão inativação do X, à RTT variante, até RTT clássica. Enquanto, nos homens, o espectro pode ir de encefalopatia grave de início neonatal com mortalidade precoce, à sinais piramidais, síndrome de parkinsonismo e macroorquidismo, até deficiência intelectual grave (KAUR; CHRISTODOULOU, 2001).
A RTT é subdivida em típica ou atípica. A RTT típica é caracterizada por uma fase regressiva, em que ocorre perda das habilidades manuais intencionais adquiridas, perda da linguagem falada adquirida, anormalidades da marcha e movimentos estereotipados das mãos. Essa fase é seguida de recuperação ou estabilização. Essas alterações não podem ser causadas por lesão cerebral secundária a trauma (peri ou pós-natal), doença neurometabólica ou infecção grave que causa problemas neurológicos. E não podem suceder de um desenvolvimento psicomotor grosseiramente anormal nos primeiros 6 meses de vida. Enquanto a RTT atípica pode não ter todas essas alterações citadas, ou pode ter outras manifestações adicionais (NEUL, 2010).
Durante a fase regressiva da RTT, ocorre uma interrupção da interação social (entre 1 e 4 anos de idade) e durante a fase de recuperação ou estabilização pode ocorrer uma melhora na comunicação social (AMERICAN PSYCHIATRIC ASSOCIATION, 2013).
95% dos pacientes com RTT clássica sobrevivem até os 20 anos, 80% até os 35 anos e cerca de 70% até aos 50 anos. Para garantir a longevidade, os pacientes com esse transtorno necessitam de uma supervisão vigorosa e adequada de sua saúde, incluindo estado nutricional, problemas gastrointestinais, escoliose, risco de aspiração e epilepsia, por parte dos pais e dos médicos que os acompanham. Além disso, o tratamento precisa perdurar durante toda a vida, e não apenas durante os primeiros anos de diagnóstico, garantindo, assim, qualidade de vida ao indivíduo (TARQUINIO, 2015).
A fisioterapia e a terapia ocupacional são fundamentais no tratamento desse distúrbio, trazendo benefícios sociais, psicológicos e também nas funções sensoriais e motoras. Diferentes modalidades, que abrangem todas as alterações no paciente com RTT, podem ser exploradas, como a hidroterapia, a equoterapia, a fonoaudiologia e a musicoterapia (SILVA; PASSOS; PARREIRA, 2016).
Diante da complexidade desse cenário genético, das projeções de sobrevida e da implicação clínica, necessitando de cuidados abrangentes e continuados, destaca-se a importância da atenção ao tema. O presente trabalho busca, por meio de uma revisão integrativa, esclarecer, de maneira abrangente e embasada, a mutação no gene MECP2, uma das mais relatadas entre as alterações genéticas no TEA, e sua repercussão na clínica dos indivíduos, constituindo a RTT, contribuindo para compreensão genética e para avanços na abordagem clínica e terapêutica desses pacientes.
2 DESENVOLVIMENTO
2.1 Metodologia
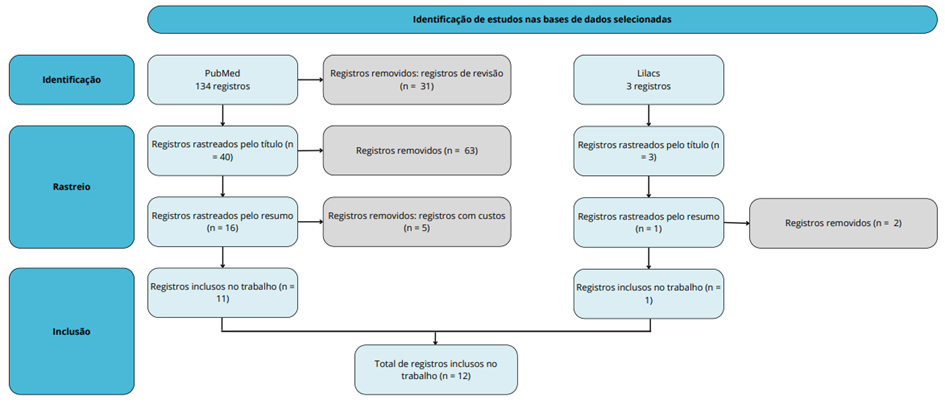
Foi realizada uma revisão integrativa mediante a seleção de artigos, demonstrada no fluxograma abaixo (Fluxograma 1). A triagem dos trabalhos se deu a partir do levantamento bibliográfico publicado nas seguintes bases de dados: National Library of Medicine (PubMed MEDLINE) e Literatura Latino-Americana e do Caribe em Ciências da Saúde (LILACS), no período de 2018 a 2023, nos idiomas inglês e espanhol.
Os artigos desse período foram selecionados a partir da busca nas bases de dados citadas pelos seguintes descritores em inglês: “autism”, “ASD”, “genetics” e “MECP2”, e em espanhol: “autismo”, “TEA” “genética” e “MECP2”, combinados entre si pelo operador booleano “AND” e “OR” com o intuito de refinar a busca. Artigos de revisão, artigos com custos, artigos em outros idiomas e títulos que não contém um dos descritores acima foram os critérios de exclusão pré-estabelecidos.
Fluxograma 1 – Ilustração das três etapas utilizadas para a seleção dos artigos: identificação, rastreio e inclusão.
2.2 Resultados e discussão
A aplicação da metodologia resultou na seleção de 12 artigos, sendo 9 deles estudos experimentais com animais que resultaram na clínica ou comportamento semelhantes a RTT, e outros 3 artigos voltados para a análise, a partir da biologia molecular e da biomedicina, de indivíduos e animais portadores da RTT. O quadro abaixo mostra os artigos, em sequência cronológica, e a abordagem utilizada em cada um deles.
Quadro 1 – Esboço do desenho experimental e análise metodológica dos artigos escolhidos.
Estudos Delineamento Abordagem/técnica Lee, Kim e Han (2018) Estudo experimental O estudo envolveu a utilização de ratos machos C57BL/6 com sete semanas de idade. Foram realizadas injeções estereotáxicas de siRNA para modificar genes específicos, associados ao autismo, em áreas cerebrais dos ratos. Os efeitos das injeções de siRNA foram examinados por meio de testes comportamentais. Zhong et al. (2019) Estudo experimental O estudo utilizou camundongos knockin fosfo-mutantes MECP2 para investigar os efeitos da fosforilação da proteína MECP2 no sistema nervoso. Foram realizadas as análises de Western blot para examinar a fosforilação do S421 da MECP2. Além disso, foram isolados e cultivados os neurônios do hipocampo e foi realizado eletrofisiologia, testes comportamentais e teste de condicionamento. Bertoldi et al. (2019) Estudo experimental Foram utilizados camundongos mutantes MECP2 como modelos para investigar os efeitos da deficiência de MECP2 em eventos de plasticidade sináptica, maturação neuronal e comportamento. Isso envolveu o uso de paradigmas de atividade in vivo, ensaios neurobiológicos, estudo de células granulares nascidas em adultos e testes comportamentais. Aron et al. (2019) Estudo observacional de coorte O estudo incluiu mulheres com suspeita ou diagnóstico clínico da RTT. Informações clínicas foram coletadas através de questionários que abordaram história familiar, história perinatal e critérios diagnósticos da RTT. O gene MECP2 foi sequenciado para detectar variantes, e foram realizadas análises de deleções ou duplicações em MECP2 usando uma técnica de amplificação por sonda dependente de ligação múltipla (MLPA). Li et al. (2020) Estudo experimental Foi investigado a capacidade de proteínas mutantes MECP2-GFP WT e R168X em particionar condensados de heterocromatina por meio de superexpressão em células. Além disso, utilizaram técnicas como citometria de fluxo, Western blot, geração de camundongos quiméricos, microscopia, ensaios in vitro, ensaios FRAP in vitro e purificação de proteínas recombinantes. Pejhan, Bigio e Rastegar (2020) Estudo observacional O estudo coletou dados observacionais de amostras de autópsia de pacientes com RTT. O estudo envolveu a avaliação dos níveis de transcrição e proteína de componentes da rede reguladora de feedback MECP2-BDNF-miR132 em diferentes regiões do cérebro humano, utilizando técnicas como RT-PCR em tempo real, Western blot e ELISA para analisar as amostras. Lu et al (2020) Estudo experimental No estudo foram utilizados camundongos para investigar os efeitos da edição epigenética na expressão do gene MECP2 e no comportamento dos animais. O estudo envolveu diversas etapas, incluindo a construção de plasmídeos, cultivo de células, análise de clivagem e sequenciamento de DNA, coloração imunofluorescente, análise de proteínas, medição de expressão gênica, injeção de zigotos e transferência de embriões, testes comportamentais em camundongos e sequenciamento para avaliar a metilação do DNA. Cai et al. (2020) Estudo experimental No estudo, participaram 5 macacos superexpressos de MECP2 (Maca fascicularis) e 20 macacos do tipo selvagem (WT). Os macacos foram submetidos a três tarefas comportamentais: observação em gaiolas, teste de separação de pares e tarefas de aprendizagem reversa. Também foram realizados experimentos EEG e de ressonância magnética. Além disso, os dados do transcriptoma de estudos anteriores foram reanalisados. Zhou et al. (2022) Estudo experimental e observacional Os pesquisadores utilizaram uma abordagem chamada Identificação de Biotina Dependente de Proximidade seguida de Espectrometria de Massa (BioID-MS) para investigar as interações do MECP2 com proteínas no contexto da cromatina, isso permitiu que fosse identificado o complexo de cromatina TCF20 como um interator do MECP2. Dessa forma, foi conduzido estudos bioquímicos, morfológicos, comportamentais e de expressão gênica. Também foi examinado pacientes humanos com mutações em genes relacionados a esses complexos de proteínas para verificar se compartilhavam características da RTT. Xu et al. (2022) Estudo experimental Neste estudo, foram criados ratos transgênicos com duplicação do gene MECP2. Foi realizado análises de expressão de proteínas, testes comportamentais e exames de ressonância magnética em diferentes faixas etárias desses ratos. O estudo visava compreender os efeitos da duplicação do gene MECP2 em termos de características fisiológicas, comportamentais e cerebrais. Mok et al. (2022) Estudo experimental Este estudo analisou os fenótipos de neurônios relacionados à RTT, um distúrbio do desenvolvimento neurológico. Utilizou modelos de neurônios derivados de células-tronco humanas para comparar células saudáveis com células afetadas por mutações no gene MECP2. Foi utilizado registros eletrofisiológicos e matrizes de microeletrodos para investigar as alterações elétricas neronasi que afetam a atividade de redes neurais. Pepe et al. (2023) Estudo experimental Neste estudo, foram utilizados camundongos mutantes heterozigotos MECP2+/- e camundongos do tipo selvagem WT. Os camundongos foram alojados individualmente em condições controladas de temperatura e umidade com ciclo claro/escuro de 12 horas. A integridade da barreira hematoencefálica (BHE) foi avaliada com FITC-albumina e imunohistoquímica. Foram realizados imunoblottings em diferentes regiões cerebrais para avaliar proteínas relacionadas à BHE. A extração de RNA e a transcrição reversa qPCR foram usadas para análise de expressão gênica.
Os estudos de Cai et al. (2020) e Xu et al. (2022) investigaram os efeitos da duplicação do gene MECP2 em modelos animais (primatas e ratos, respectivamente). Ambos os estudos encontraram mais resultados que corroboram e ampliam a compreensão dos impactos dessa duplicação em termos de expressão gênica e conectividade neural, e como esta afeta, principalmente, o comportamento e até a estrutura cerebral.
O estudo de Cai et al. (2020) investigou os efeitos da duplicação do gene MECP2 na conectividade neural de um modelo de primata. Os resultados sugerem que essa duplicação afeta tanto a conectividade temporal quanto a conectividade espacial, afetando várias redes cerebrais, incluindo a rede fronto-parieto-occipital, a rede pré-frontal e a cingulada. Além disso, a conectividade anormal observada nos macacos transgênicos se assemelha às observadas em um grupo de pessoas com autismo. A pesquisa destacou a importância dessa conexão anormal para o diagnóstico preciso de distúrbios do espectro autista e potencialmente para o desenvolvimento de intervenções comportamentais destinadas a melhorar a locomoção atípica e a comunicação social.
Além disso, os pesquisadores observaram que a duplicação do gene MECP2 estava associada a um módulo de genes que incluía genes relacionados à função GABA e estava associada a transtornos de humor. A análise da expressão gênica revelou que muitos genes ligados ao MECP2 eram regulados de maneira significativa nos macacos transgênicos. Além disso, os macacos transgênicos apresentaram déficits comportamentais, como locomoção repetitiva e aumento da taxa de erros regressivos em tarefas de aprendizagem. Essas anormalidades comportamentais estavam correlacionadas com as alterações na conectividade neural.
Da mesma forma, a pesquisa de Xu et al. (2022) também examinou os efeitos da duplicação do gene MECP2, porém em ratos. Os pesquisadores observaram um aumento significativo na expressão da proteína MECP2 em várias áreas do cérebro, incluindo o córtex cerebral e o hipocampo. Além disso, os ratos com a duplicação do gene MECP2 apresentaram um crescimento mais lento do peso corporal e uma taxa de sobrevivência reduzida. Em termos de comportamento, esses ratos mostraram déficits significativos, incluindo menor atividade locomotora e menor preferência por interações sociais em testes comportamentais, semelhantes aos resultados encontrados por Cai et al. (2020).
Os resultados também revelaram mudanças nas redes cerebrais subjacentes a esses déficits. Os pesquisadores identificaram anormalidades na atividade funcional e estrutura cerebral, com foco em áreas como o córtex pré-frontal medial dorsal (dmPFC), córtex retroesplenial (RSP), córtex motor e outras regiões do cérebro. Essas mudanças funcionais e estruturais foram associadas aos déficits sociais e motores observados nos ratos MECP2-DP. Além disso, as análises longitudinais indicaram que as alterações no desenvolvimento cerebral induzidas pela duplicação do MECP2 não eram significantes durante a adolescência dos ratos, sugerindo que essas alterações podem ocorrer em fases anteriores do desenvolvimento.
A pesquisa de Pejhan, Bigio e Rastegar (2020), assim como a de Xu et al. (2022), incluiu uma análise histopatológica da estrutura cerebral dos cérebros de pacientes com RTT e controles, com foco nas regiões frontal, hipocampo e cerebelo. Foram utilizadas colorações de hematoxilina e eosina para avaliar possíveis anormalidades anatômicas em níveis patológicos. Não foram observadas características notáveis, como neurônios mortos ou inflamação, em exames microscópicos. No entanto, algumas mudanças sutis foram detectadas, incluindo neurônios piramidais menores e maior densidade celular no hipocampo em pacientes com a mutação T158M, bem como um giro denteado mais fino e focalmente hipocelular no hipocampo de pacientes com a mutação A201V.
A partir de diferentes intervenções, os estudos de Lee, Kin e Han (2018), Zhong et al. (2019) e Lu et al. (2020), analisaram alterações em MECP2 e suas consequências clínicas, resultando na RTT. Todos observaram mudanças comportamentais semelhantes às do autismo, assim como Cai et al. (2020) e Xu et al. (2022), destacando o papel crucial do MECP2 nesse contexto.
Os resultados do estudo de Lee, Kin e Han (2018) indicam que a inibição mediada por siRNA de genes relacionados ao TEA, como SHANK3, NLGN3, FMR1, MECP2 e TSC1, no corpo estriado dorsal de camundongos produziu uma variedade de comportamentos semelhantes aos do autismo, variando de leves a graves. Os camundongos submetidos a essa inibição apresentaram mudanças no comportamento, incluindo comportamento de limpeza, escavação, preferência social por novidades, preferência por novos objetos e locomoção, que são relevantes para a compreensão do TEA.
No entanto, o grau de mudança comportamental variou de acordo com o gene inibido. Os resultados destacam que o knockdown de MECP2 e TSC1 no corpo estriado dorsal produziu as mudanças comportamentais mais pronunciadas e robustas, sugerindo um papel fundamental desses genes nesse contexto. Além disso, o estudo demonstrou que esses comportamentos autistas induzidos pela inibição de MECP2 ou TSC1 puderam ser revertidos ou modulados por intervenções farmacológicas, como D-cicloserina, fenobam, SCH23390 e ecopipam, que afetam os sistemas de neurotransmissores glutamato e dopamina no corpo estriado dorsal.
O estudo Zhong et al. (2019) também envolveu a geração de camundongos com uma mutação na proteína MECP2, especificamente no aminoácido 421, onde a serina foi substituída por ácido glutâmico para imitar a fosforilação. Foi observada a fosforilação da MECP2 na linhagem MECP2 S421E. No entanto, o nível total de proteína MECP2 permaneceu semelhante entre os camundongos do tipo selvagem e MECP2 S421E. Camundongos MECP2 S421E apresentaram déficits na aprendizagem e memória do medo, bem como redução na densidade de sinapses excitatórias em comparação com camundongos do tipo selvagem.
Além disso, investigou-se como a imitação da fosforilação em S421 afeta a escala sináptica. Os resultados mostraram que os camundongos MECP2 S421E apresentaram um déficit na ampliação das sinapses excitatórias induzido por TTX, mas não na redução induzida pela bicuculina, em comparação com os camundongos do tipo selvagem. Outro aspecto do estudo envolveu camundongos MECP2 S80A, nos quais a serina no aminoácido 80 foi mutada. Esses camundongos demonstraram prejuízo na aprendizagem espacial e memória. Além disso, nas células progenitoras neurais, houve um aumento na proliferação e uma diminuição na diferenciação nas células MECP2 S80A.
Foi investigado, por meio do estudo de Lu et al. (2020), o papel da metilação do promotor do gene MECP2 na patogênese do TEA. Os pesquisadores utilizaram uma técnica de edição de metilação de DNA baseada em dCas9 para aumentar seletivamente a metilação no promotor MECP2. A metilação direcionada do promotor MECP2 levou a um aumento na metilação do promotor, resultando na regulação negativa da expressão do gene MECP2. Além disso, a metilação do DNA foi analisada em várias regiões fora do alvo, e apenas algumas regiões mostraram um leve aumento na metilação.
Os pesquisadores também injetaram os vetores de metilação e controle em zigotos de camundongos e observaram uma maior metilação do DNA em várias regiões do genoma. Camundongos machos no grupo de metilação apresentaram comportamentos semelhantes aos do TEA, como dificuldades de interação social, comportamentos repetitivos, redução na locomoção e aumento da ansiedade. Além disso, os pesquisadores realizaram injeções de vetores de metilação no hipocampo de camundongos adultos e observaram que a metilação direcionada do promotor MECP2 no hipocampo também resultou em alterações comportamentais semelhantes aos sintomas do TEA.
O estudo de Bertoldi et al. (2019) investigou os efeitos de convulsões em camundongos com mutações no gene MECP2, associado à RTT. Para realizar o estudo, camundongos MECP2-308 (MUT) e seus irmãos de ninhada de tipo selvagem (WT) foram injetados com ácido caínico (KA) para induzir convulsões, a fim de ativar a atividade neuronal. Resultados mostraram que os camundongos MUT responderam ao KA com atividade convulsiva semelhante aos WT, indicando que a ativação neuronal não estava prejudicada em camundongos com mutações no gene MECP2. Entretanto, quando avaliaram o tamanho do trato de fibras no hipocampo (IPT) duas semanas após a administração de KA, observaram que apenas os camundongos WT apresentaram um aumento significativo, enquanto os MUT não mostraram alterações, sugerindo que a mutação de MECP2 afeta negativamente a capacidade de resposta plástica do IPT às convulsões induzidas por KA, afetando a maturação de neurônios granulares e a expressão de moléculas de orientação axonal.
Além disso, a deficiência no fator neurotrófico derivado do cérebro (BDNF), um fator de crescimento importante na plasticidade neuronal, também foi observada nos camundongos MUT. Essas descobertas fornecem informações sobre distúrbios neurológicos associados à mutação do gene MECP2.
No estudo de Zhou et al. (2022), os pesquisadores investigaram a interação entre a proteína MECP2 e o complexo TCF20 no cérebro de camundongos. Eles descobriram que TCF20, PHF14 e outras proteínas fazem parte de um complexo de ligação à cromatina que interage com o MECP2. Mutações no MECP2 afetam essa interação, especialmente em relação à RTT. TCF20 e MECP2 foram observados como coexpressos em neurônios de camundongos e desempenham um papel na regulação da formação de sinapses. A análise da expressão gênica indicou padrões de expressão espacial e temporal semelhantes para TCF20 e MECP2.
Foi descoberto, também, que a superexpressão de MECP2 aumentou o número de sinapses em neurônios, mas a redução de TCF20 em neurônios com superexpressão de MECP2 restaurou os níveis normais de sinapses. TCF20 também estava envolvido na regulação do gene BDNF, que afeta a densidade de sinapses em neurônios.
Os camundongos com haploinsuficiência de TCF20 apresentaram déficits de aprendizagem, memória e comportamento semelhantes à ansiedade, que foram revertidos pela redução de TCF20 em camundongos com superexpressão de MECP2. A sobreposição de déficits comportamentais sugeriu uma interação entre TCF20 e MECP2 na regulação de vias neuronais comuns. A redução de TCF20 aliviou alguns déficits comportamentais em camundongos com superexpressão de MECP2, e a imunoprecipitação de MECP2 revelou um maior recrutamento de TCF20 e PHF14 em camundongos com superexpressão de MECP2. Além disso, mutações no gene PHF14 foram associadas a um fenótipo neurológico semelhante ao autismo e à RTT.
O trabalho de Pejhan, Bigio e Rastegar (2020) também investigou a expressão de vários genes e proteínas em diferentes regiões do cérebro de pacientes com RTT e controles. Observou-se que os níveis de transcrição das isoformas MECP2/MECP2E1 e E2, BDNF/BDNF e miR132 foram comprometidos em pacientes com RTT, embora essa alteração não tenha sido uniforme em todas as partes do cérebro. Os resultados também indicaram que os níveis de proteína MECP2E1/E2-BDNF não seguiram os mesmos padrões de transcrição, sugerindo que a regulação da homeostase MECP2E1/E2-BDNF-miR132 pode não ser consistente em diferentes regiões do cérebro humano.
Os artigos de Bertoldi et al. (2019), Zhou et al. (2022) e Pejhan, Bigio e Rastegar (2020) compartilham o foco no gene MECP2 e suas implicações em distúrbios neurológicos, especialmente na RTT. Embora cada estudo aborde aspectos diferentes e utilize abordagens específicas, a convergência nos temas de MECP2, plasticidade cerebral, alterações nas redes neurais e distúrbios neurológicos fornece uma base sólida para uma compreensão das implicações neurológicas dessas mutações. Além disso, os estudos mencionam o fator neurotrófico derivado do cérebro (BDNF) em associação com mutações no gene MECP2. A deficiência no BDNF é sugerida como uma contribuinte para os déficits neuronais e comportamentais observados. Bertoldi et al. (2019) observam essa deficiência após convulsões induzidas por KA, enquanto Zhou et al. (2022) relacionam a regulação do gene do BDNF por TCF20 e seu papel na densidade de sinapses.
Complementando esses estudos, Pepe et al. (2023) investigaram a permeabilidade da barreira hematoencefálica (BHE) em camundongos com RTT. A análise revelou um aumento na permeabilidade da BHE em camundongos RTT em comparação com camundongos do tipo selvagem (WT). Isso foi evidenciado pela presença de fluorescência difusa de FITC-albumina nos tecidos cerebrais de camundongos RTT, enquanto camundongos WT não apresentaram sinal de extravasamento de albumina FITC.
A expressão de proteínas-chave da junção estreita (TJ), como claudina-5 e ocludina, diminuiu em várias regiões cerebrais de camundongos RTT, indicando uma possível correlação entre a diminuição na expressão dessas proteínas e a quebra da BHE. Além disso, vários genes envolvidos na estrutura e função da BHE, como CLDN3, CLDN12, JAM2, AQP4, MPDZ e MMP9, mostraram níveis de expressão alterados em camundongos RTT em comparação com controles WT, sugerindo que múltiplos fatores podem estar envolvidos na disfunção da BHE. A interrupção da BHE tem sido associada a várias condições neurológicas, e este estudo fornece evidências iniciais da perturbação da BHE na RTT. Essas descobertas podem abrir caminho para o desenvolvimento de biomarcadores e terapias potenciais para a RTT, considerando o impacto da disfunção da BHE no metabolismo cerebral, nas redes neurais e na resposta inflamatória.
Mok et al. (2022) forneceram achados valiosos sobre as diferenças nos neurônios associadas à RTT, destacando a complexidade dessa condição neurológica e contribuindo para uma compreensão mais aprofundada de suas bases celulares e moleculares. A pesquisa investigou neurônios como um modelo celular para a RTT, focando em dois grupos principais: neurônios nulos MECP2, associados à RTT, e neurônios com uma mutação missense chamada CLT L124W. O estudo analisou a proteína MECP2 e suas mutações, com ênfase na mutação L124W. Foram utilizadas linhagens celulares isogênicas para comparar o impacto da mutação L124W com outras mutações na arquitetura da cromatina. Observou-se que a mutação L124W causava rupturas limitadas na heterocromatina e diferenças na dinâmica de ligação em comparação com o tipo selvagem (WT).
Além disso, foram geradas linhagens de células-tronco pluripotentes induzidas (iPSCs) com a mutação L124W, usadas para derivar neurônios excitatórios. Os níveis de proteína MECP2 nessas células foram semelhantes aos das células WT. No entanto, os neurônios nulos MECP2 apresentaram várias diferenças em relação aos controles isogênicos, como um potencial de membrana em repouso despolarizado, maior resistência de entrada e uma diminuição na capacitância celular. Além disso, foi demonstrado alterações nas propriedades da membrana ativa, com reduções nas correntes de sódio (Na+) e potássio (K+) dependentes de voltagem, indicando disfunção no disparo do potencial de ação e hipoatividade sináptica excitatória.
A análise da morfologia revelou que os neurônios nulos MECP2 tinham tamanho de soma e comprimento total dos dendritos reduzidos, associados à microcefalia. Também apresentaram menor complexidade na ramificação dendrítica e menor densidade de sinapses excitatórias. Em experimentos de redes neuronais, observou-se um padrão de atividade sincronizada anômala nas culturas de neurônios RTT, com uma redução na frequência de rajadas de rede em comparação com os controles isogênicos. Modelos de simulação sugeriram que essas mudanças estavam relacionadas a alterações nas correntes de sódio e potássio, destacando o papel das propriedades intrínsecas dos neurônios individuais nessas alterações.
Surpreendentemente, os neurônios CLT L124W com a mutação missense, compartilharam algumas características com os neurônios nulos MECP2, como aumento da resistência de entrada e correntes de sódio e potássio prejudicadas. No entanto, eles não demonstraram as mesmas deficiências em potencial de ação e função sináptica.
A pesquisa de Li et al (2020) descreve uma série de experimentos e procedimentos realizados no contexto de pesquisa em biologia molecular e biomedicina que, em adição, contribui na compreensão da RTT. Os principais pontos incluem a superexpressão de proteínas mutantes MECP2-GFP WT e R168X em células para investigar sua capacidade de particionar condensados de heterocromatina, o uso de citometria de fluxo para avaliar os níveis relativos de expressão de proteínas marcadas endógenas nas células, a realização de Western blot, uma técnica utilizada para analisar e quantificar proteínas, para confirmar a expressão das proteínas MECP2-GFP WT e R168X, a geração de camundongos quiméricos marcados com MECP2-GFP endógenos por meio da injeção de células-tronco embrionárias em embriões de camundongo, a microscopia de imunofluorescência em fatias cerebrais para estudar o tamanho e o número de condensados de heterocromatina em neurônios, a realização de ensaios de gotículas in vitro para investigar as propriedades físico-químicas de proteínas associadas à condensados, a realização de ensaios de gotículas FRAP in vitro para avaliar a dinâmica das gotículas e a purificação de proteínas recombinantes para uso em experimentos.
Esses procedimentos fazem parte de uma pesquisa abrangente com o objetivo de compreender como as proteínas interagem em condensados celulares e sua relação com a biologia molecular e celular. Além disso, o estudo investigou as características do fragmento MECP2 Mini e seu papel na formação de condensados, com foco na RTT.
Os resultados do estudo oferecem informações sobre as interações entre proteínas em condensados celulares e como as mutações podem impactar essas interações. Especialmente em relação à RTT, a descoberta fundamental deste estudo é que as mutações prejudicam a capacidade do MECP2 de formar condensados celulares, indicando que a desestabilização desses condensados pode ser uma consequência comum das mutações em pacientes com RTT. Além disso, Zhou et al. (2022) apoiam o resultado, que sugere que o MECP2 desempenha um papel na segregação entre heterocromatina e eucromatina por meio de suas propriedades de partição condensada, quando sugere que um complexo de ligação à cromatina interage com o MECP2.
O estudo de Aron et al. (2019) foi em contrapartida dos demais estudos, sugerindo que a RTT pode ter outras causas genéticas além da associação com MECP2. Iniciou selecionando mulheres chilenas com suspeita clínica de RTT que foram examinadas para variantes patogênicas no gene MECP2 e, então, foram coletadas informações clínicas por meio de questionários. Dos 14 pacientes incluídos, 10 foram diagnosticados com RTT clássico e 4 com RTT atípico. Variantes patogênicas no gene MECP2 foram encontradas em 8 dos 14 pacientes, com a maioria delas localizadas nos éxons 3 e 4. Além disso, uma deleção no éxon 3 foi detectada em 2 pacientes. No entanto, 5 pacientes não apresentaram variantes patogênicas identificadas, sugerindo que outras causas genéticas podem estar envolvidas nos casos de RTT sem variantes no gene MECP2. Esse estudo contribuiu para o diagnóstico molecular da RTT no Chile.
3 CONSIDERAÇÕES FINAIS
As mutações no MECP2 levam à alterações comportamentais, déficits na aprendizagem, na memória do medo, e nas sinapses excitatórias. Além disso, causam prejuízo na aprendizagem espacial e memória. Ainda, afetam a resposta do hipocampo a convulsões, com implicações para a plasticidade neuronal e a geração de novos neurônios.
Foi observado que, na RTT, ocorre um aumento na permeabilidade da barreira hematoencefálica, impactando o metabolismo cerebral, as redes neurais e a resposta inflamatória.
Em síntese, esses estudos não apenas colaboram para o conhecimento da RTT em níveis moleculares, celulares e comportamentais, mas também destacam a complexidade e a interligação de diferentes fatores que contribuem para essa síndrome neuropsiquiátrica. A confluência de resultados em diferentes níveis de análise fornece uma visão integrativa das bases neurobiológicas da RTT.
REFERÊNCIAS
AMERICAN PSYCHIATRIC ASSOCIATION. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-V). Arlington, VA: American Psychiatric Association, 2013.
ARON, C. W. et al. Síndrome de Rett: Análisis molecular del gen MECP2 en pacientes chilenas. Revista Chilena de Pediatría, Santiago, v. 90, n. 2, p. 152-156, 2019.
BERTOLDI, M. L. et al. MeCP2 Deficiency Disrupts Kainate-Induced Presynaptic Plasticity in the Mossy Fiber Projections in the Hippocampus. Frontiers in cellular neuroscience, v. 13, p. 286, 2019.
CAI, D. C. et al. MECP2 Duplication Causes Aberrant GABA Pathways, Circuits and Behaviors in Transgenic Monkeys: Neural Mappings to Patients with Autism. The Journal of neuroscience: the official journal of the Society for Neuroscience, v. 40, n. 19, 2020.
CHIDAMBARAM, S. B. et al. Protein Nutrition in Autism. Advances in neurobiology, v. 24, p. 573-586, 2020.
CHOI, M. et al. Autistic-like social deficits in hippocampal MeCP2 knockdown rat models are rescued by ketamine. BMB reports v. 55, n. 5, p. 238-243, 2022.
KAUR, S. CHRISTODOULOU, J. MECP2 Disorders. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; October 3, 2001.
LEE, Y.; KIM, H.; HAN, P. L. Striatal Inhibition of MeCP2 or TSC1 Produces Sociability Deficits and Repetitive Behaviors. Experimental neurobiology, v. 27, n. 6, p. 539–549, 2018.
LI, C. H. et al. MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature, v. 586, n. 7829, p. 440-444, 2020.
LU, Z. et al. Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell death & disease, v. 11, n. 2, 2020.
MAENNER, MJ. et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years — Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2018. MMWR Surveill Summ. 2021.
MOK, R. S. F. et al. Wide spectrum of neuronal and network phenotypes in human stem cell-derived excitatory neurons with Rett syndrome-associated MECP2 mutations. Translational psychiatry, v. 12, n. 1, 2022.
NEUL, J. L. et al. Rett syndrome: revised diagnostic criteria and nomenclature. Annals of neurology, v. 68, n. 6, 2010.
PEJHAN, S.; DEL BIGIO, M. R.; RASTEGAR, M. The MeCP2E1/E2-BDNF-miR132 Homeostasis Regulatory Network Is Region-Dependent in the Human Brain and Is Impaired in Rett Syndrome Patients. Frontiers in cell and developmental biology, v. 8, 2020.
PEPE, G. et al. Blood-Brain Barrier Integrity Is Perturbed in a Mecp2-Null Mouse Model of Rett Syndrome. Biomolecules, v. 13, n. 4, 2023.
SILVA, N.; PASSOS, X.; PARREIRA, S. Síndrome de Rett: uma revisão da literatura. Journal of the Health Sciences Institute, 2016.
STYLES, M. et al. Risk factors, diagnosis, prognosis and treatment of autism. Front. Biosci. (Landmark Ed) 2020.
TA, D. et al. A brief history of MECP2 duplication syndrome: 20-years of clinical understanding. Orphanet journal of rare diseases v. 17, n. 1: 131, 2022.
TARQUINIO, D. C. et al. The Changing Face of Survival in Rett Syndrome and MECP2-Related Disorders. Pediatric neurology, v. 53, n. 5, p. 402-411, 2015.
THAPAR, A.; RUTTER, M. Genetic Advances in Autism. Journal of autism and developmental disorders, v. 51, n. 12, p. 4321-4332, 2021.
XU, M. et al. Aberrant brain functional and structural developments in MECP2 duplication rats. Neurobiology of disease, v. 173, 2022.
ZEIDAN, J. et. al. Global prevalence of autism: A systematic review update. Autism research : official journal of the International Society for Autism Research, v. 15, n. 5, p. 778-790, 2022.
ZHONG, X. et al. Regulation of neural differentiation, synaptic scaling and animal behavior by MeCP2 phosphorylation. Neurobiology of learning and memory, v. 165, 106859, 2019.
ZHOU, J. et al. Disruption of MeCP2-TCF20 complex underlies distinct neurodevelopmental disorders. Proceedings of the National Academy of Sciences of the United States of America, v. 119, n. 4, 2022.
¹Acadêmica do Curso de Medicina, UNICESUMAR – Universidade Cesumar, Campus Maringá. Programa Voluntário de Iniciação Científica (PVIC/ Unicesumar). abssbia@hotmail.com
²Acadêmica do Curso de Medicina, UNICESUMAR – Universidade Cesumar, Campus Maringá. Programa Voluntário de Iniciação Científica (PVIC/ Unicesumar). analuforti.neves@outlook.com
³Orientadora, Medicina, Unicesumar, clarissa.torresan@docentes.unicesumar.edu.br