REGISTRO DOI: 10.69849/revistaft/ar10202412121031
Gabriel Cardoso Nicolai;
Orientador: Dra. Ana Carolina Ignácio Colussi Bressan.
RESUMO
As microangiopatias trombóticas (MATs) caracterizam-se por oclusão microvascular generalizada, que culmina em anemia hemolítica microangiopática, trombocitopenia e lesão orgânica. Fenotipicamente podem manifestar-se como Púrpura Trombocitopênica Trombótica e Síndrome Hemolítica-Urêmica, condições clinicamente graves e com elevada morbimortalidade.
O objetivo do trabalho é relatar o caso de uma paciente jovem, admitida no pronto atendimento do Complexo Hospitalar Prefeito Edvaldo Orsi, na cidade de Campinas/SP, com quadro de dispneia e hipertensão arterial secundárias à hipervolemia, apresentando disfunção renal aguda grave e necessidade de suporte dialítico em caráter de urgência. Após avaliação dos exames laboratoriais aventou-se a hipótese de Síndrome Hemolítico-Urêmica (SHU) que foi confirmada com biópsia renal realizada em serviço externo posteriormente. As informações contidas neste relato foram obtidas por meio de revisão de prontuário, entrevista com equipe médica, registro dos exames complementares e revisão de literatura.
As limitações de recursos do serviço no qual a paciente foi admitida já com disfunção renal instalada, somada ao fato de que o Eculizumabe, anticorpo monoclonal usado para tratamento de casos de Síndrome Hemolítico-Urêmica atípica (SHUa) não é disponibilizado pelo Ministério da Saúde no Brasil para tratamento de SHU culminou, respectivamente, no diagnóstico tardio e na impossibilidade de tratamento adequado. Paciente evoluiu com Doença Renal Crônica terminal e necessidade de terapia renal substitutiva crônica intermitente.
Palavras-chave: Microangiopatia trombótica, Síndrome Hemolítico-Urêmica, eculizumabe, SHU atípica, SHU atípica mediada por complemento.
ABSTRACT
Thrombotic microangiopathies (MATs) are characterized by generalized microvascular occlusion, which culminate in microangiopathic hemolytic anemia, thrombocytopenia and organ damage. Phenotypically, they may manifest as Thrombotic Thrombocytopenic Purpura and Hemolytic-Uremic Syndrome, clinically serious conditions with high morbidity and mortality.
The objective of this study is to report the case of a young patient, admitted to the emergency department of the Complexo Hospitalar Prefeito Edvaldo Orsi, in the city of Campinas/SP, with dyspnea and arterial hypertension secondary to hypervolemia, associated with severe acute renal dysfunction and need for emergency dialysis. After evaluating the laboratory tests, the hypothesis of Hemolytic-Uremic Syndrome (HUS) was suggested, which was later confirmed with a renal biopsy performed in an outpatient service. The information contained in this report was transmitted through a review of medical records, an interview with the medical team, registration of complementary exams and a review of the literature.
The resource restrictions of the service where the patient was admitted with installed renal dysfunction, added to the fact that Eculizumab, a monoclonal antibody used to treat cases of atypical Hemolytic Uremic Syndrome (aHUS) is not made available by the Ministry of Health in Brazil for HUS treatment culminated, respectively, in late diagnosis and impossibility of adequate treatment. The patient evolved with endstage Chronic Kidney Disease and required intermittent chronic renal replacement therapy.
Keywords: Thrombotic microangiopathy, Hemolytic Uremic Syndrome, eculizumab, atypical HUS, complement-mediated atypical HUS.
1. INTRODUÇÃO
As microangiopatias trombóticas (MAT) são condições patológicas que se caracterizam por oclusão microvascular generalizada decorrente da deposição de trombos plaquetários e, por conseguinte, anemia hemolítica microangiopática, trombocitopenia e lesão isquêmica nos órgãos obstruídos. (MOAKE, 2002).
A anemia hemolítica é causada pela destruição mecânica dos eritrócitos quando atravessam a microcirculação obstruída por trombos, levando a formação de esquizócitos no sangue periférico. Há aumento do LDH devido à lise celular, redução de haptoglobina (glicoproteína que se liga à hemoglobina livre durante hemólise para renovação celular) e reticulocitose decorrente da atividade hematopoiética na medula óssea. A plaquetopenia se deve ao consumo de plaquetas nos trombos ao longo da microcirculação. O teste de Coombs direto é negativo visto que há ausência de autoimunidade. (THOMPSON e KAVANAGH, 2022).
Fenotipicamente, as MATs podem manifestar-se como Púrpura Trombocitopênica Trombótica (PTT) e Síndrome Hemolítico-Urêmica (SHU) (Polito; Kirsztajn, 2010). No entanto, nas últimas décadas, esforços foram impostos para a compreensão fisiopatológica e para identificação de outras etiologias, demonstrando a abrangência da doença. (LOIRAT et al., 2015); (JOSEPH et al., 2013)
Enquanto que na PTT o acometimento orgânico é difuso, classicamente em tecido encefálico, na SHU os trombos plaquetárias ocluem predominantemente a microcirculação renal. (MOAKE, 2002).
Na PTT, há uma deficiência funcional ou quantitativa de ADAMTS-13, enzima que tem a função de degradar multímeros do fator de von Willebrand no plasma. O acúmulo desses multímeros gera oclusão de arteríolas e capilares com consequente isquemia dos órgãos-alvo. Desta forma, a avaliação laboratorial da ADAMTS-13 é critério diagnóstico para PTT e constitui avanço no diagnóstico precoce. (TONACO et al., 2010)
A SHU, por sua vez, acomete prioritariamente a microvasculatura renal. Pode ser classificada como típica, geralmente associada à infecção por Escherichia coli produtora de toxina Shiga (STEC) após quadro de diarreia sanguinolenta; ou atípica, que são todos os casos de SHU não decorrentes de STEC.
SHU atípica (SHUa) pode ser descrita como primária ou secundária. Esta última se deve a uma variedade de causas que incluem outros agentes infecciosos que não a E. coli ou Shigella, como Streptococcus pneumoniae, vírus da imunodeficiência humana (HIV) e Influenza A; malignidades; gravidez e síndrome HELLP e doenças sistêmicas, como Esclerodermia, Lúpus Eritematoso Sistêmico e Síndrome do Anticorpo Antifosfolípide. A SHUa primária pode ser familiar, visto que nenhuma causa exógena foi determinada e, durante a última década, demonstrou-se ser uma doença de desregulação do sistema complemento. (CAMPISTOL et al., 2015).
Segundo CAMPISTOL et al., (2015), o diagnóstico diferencial de SHU mediada por complemento requer a exclusão de PTT e SHU por toxina Shiga (STEC-SHU). De acordo com George, et al. (2014), sua fisiopatologia está associada a uma hiperativação da via alternativa do complemento, seja por mutações em genes regulatórios ou produção de autoanticorpos, que culmina no aumento da produção do complexo de ataque à membrana e consequente lise das células endoteliais dos vasos.
Não há biomarcadores específicos para a síndrome. O papel da medição de proteínas do complemento (C3, C4 e CH50) e anticorpos para essas proteínas ou mutações de genes do complemento não está claro. Baixas concentrações de C3 apresentam baixa sensibilidade (30% dos casos) (BAGGA et al., 2019). De acordo com Fakhouri et al. (2017), o diagnóstico não deve ser baseado na detecção de genes variantes do complemento, que são detectados em cerca de 40-60% dos casos. A ausência desses genes não exclui a possibilidade da doença.
O aumento do conhecimento da patogênese da SHUa foi acompanhado pelo surgimento do Eculizumabe, um anticorpo IgG monoclonal humanizado que se liga ao fator C5 do complemento, impedindo a formação do complexo de ataque à membrana. Estudos mostraram aumento de plaquetas e melhora da função renal já na primeira dose do medicamento (VAISBICH, et al., 2013). Segundo Walle et al. (2016), quando a terapia é iniciada em até 7 dias do início do quadro, a chance de recuperação da função renal é maior.
O diagnóstico diferencial das MAT é complexo, mas essencial para compreender as decisões de tratamento. Segundo Campistol et al. (2015), os guias de consenso para o diagnóstico diferencial foram atualizados. No entanto, evidências do mundo real sobre a atual prática de diagnóstico e tratamento não foram relatadas. A hipótese é de que existem áreas de incerteza na investigação de MAT e, como relatado por Mittal et al. (2022), a indisponibilidade de recursos diagnósticos e terapêuticos em países subdesenvolvidos e os possíveis atrasos no estabelecimento do diagnóstico final aumentam o índice de morbimortalidade posto que, segundo Loirat et al. (2015), o Eculizumabe é útil enquanto o paciente ainda apresenta lesões sugestivas de MAT e não se veem benefícios quando os rins já estão fibróticos e esclerosados.
A mortalidade pode atingir 25% dos casos, sendo que 50% dos pacientes evoluem para terapia renal substitutiva crônica em virtude da ausência de tratamento adequado (RAINA et al., 2018).
2. JUSTIFICATIVAS
O presente trabalho se trata de um relato de caso de Síndrome HemolíticoUrêmica em paciente jovem que procurou atendimento médico já com lesão renal instalada após vários dias do início dos sintomas da doença e recebeu diagnóstico tardio.
Em decorrência das limitações do serviço de saúde a qual foi admitida e à indisponibilidade do tratamento medicamentoso preconizado para casos de SHUa pela legislação brasileira, evoluiu com disfunção renal grave e cronicidade renal, permanecendo em terapia renal substitutiva.
O trabalho visa expor as disparidades entre o manejo diagnóstico e terapêutico ideal e o real, promovido em locais com precariedade de recursos, que dificulta o acolhimento deste tipo de paciente nestes serviços, determinando elevada morbidade.
3. OBJETIVOS
Descrever caso de Síndrome Hemolítico-Urêmica atípica (SHUa) acometendo paciente jovem com disfunção renal grave.
Definir microangiopatia trombótica (MAT) além dos sinais e sintomas sugestivos da doença, discutir as diferentes apresentações fenotípicas e investigação laboratorial para determinar diagnósticos diferenciais.
Expor a deficiência e limitações do serviço de saúde do Brasil no diagnóstico etiológico de MAT, bem como o difícil acesso à terapia medicamentosa preconizada para tratamento de SHUa mediada por complemento, que determinaria menor morbimortalidade aos doentes acometidos.
4. MÉTODOS
As informações contidas neste relato de caso foram obtidas por meio de revisão de prontuário, entrevista com equipe médica, registro dos exames complementares e revisão de literatura.
5. RELATO DE CASO
Paciente do sexo feminino, 17 anos, parda, sem comorbidades conhecidas, nuligesta, utilizando continuamente apenas anticoncepcional oral (diminut), deu entrada no pronto atendimento do Complexo Hospitalar Prefeito Edvaldo Orsi, Campinas/SP, referindo quadro diarreico, sem muco ou sangue há 20 dias (cerca de 4 episódios diários) associado a vômitos. Há 1 semana da admissão evoluiu com inapetência, dor abdominal difusa, tipo cólica, e edema de membros inferiores e há 2 dias da internação, apresentou quadro de dispneia em repouso, anasarca e prurido generalizado, motivo pelo qual procurou atendimento médico.
À admissão, estava descorada, hipertensa (196/108 mmHg), afebril (Tax: 36,6ºC), FC: 92bpm e SatO2: 99% em ar ambiente. Exame físico demonstrava estertores crepitantes em bases pulmonares e edema de membros inferiores.
Exames laboratoriais colhidos na primeira avaliação revelaram a presença de anemia (Hemoglobina: 5,6g/dl) normocrômica normocítica, elevação das escórias nitrogenadas (Creatinina: 14,0mg/dl e Ureia: 197mg/dl), sem alterações eletrolíticas e urinálise com hematúria e proteinúria. Apresentava contagem de plaquetas dentro da normalidade.
Foi avaliada pela equipe de nefrologia do serviço, que orientou investigação de doença glomerular e início de pulsoterapia com corticoide por hipótese diagnóstica de glomerulonefrite rapidamente progressiva, além de indicar início de terapia renal substitutiva por sintomas urêmicos e oligoanúria com hipervolemia refratária.
Tabela 1 – Achados em esfregaço de sangue periférico e EAS.

Tabela 2 – Exames laboratoriais gerais
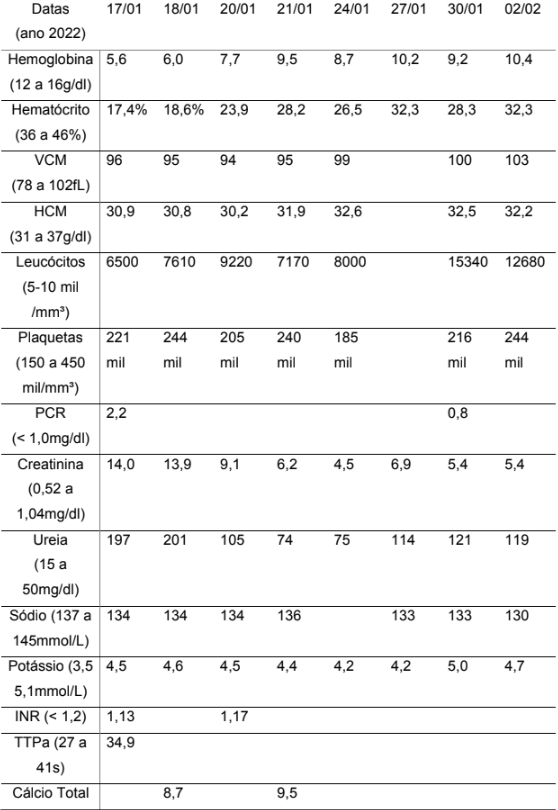
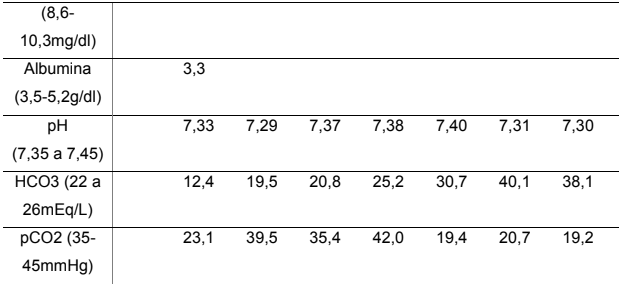
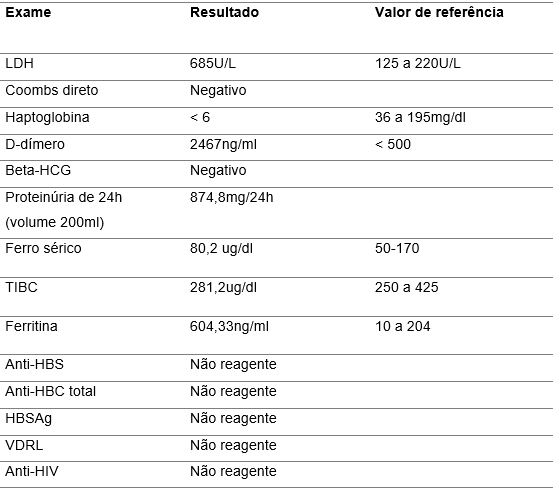
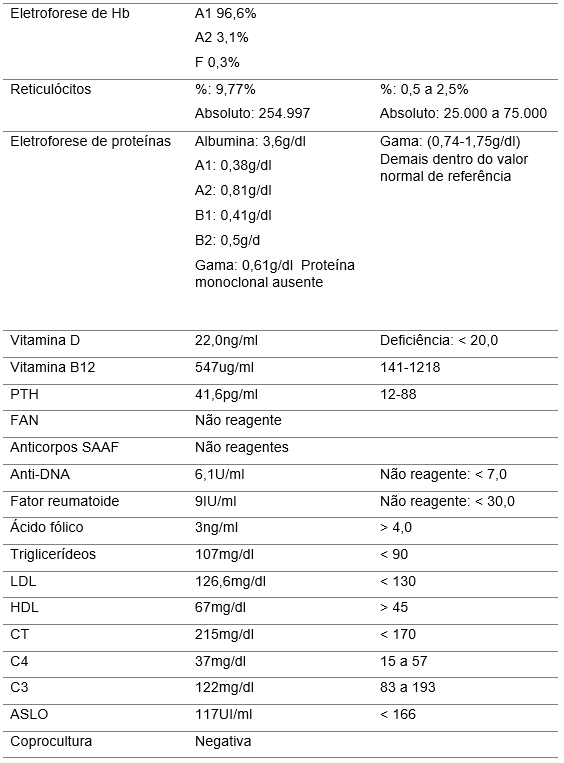
Tabela 3 – Exames investigativos específicos
Exames de imagem:
- Tomografias de tórax e abdome não apresentavam alterações, exceto por derrame pleural moderado bilateral.
- Ultrassonografia de rins e vias urinárias demonstravam rins atróficos com diferenciação córtico-medular preservada.
Aventado a hipótese de Microangiopatia Trombótica decorrente de Síndrome
Hemolítico-Urêmica visto que PTT foi afastada por apresentar pontuação baixa no PLASMIC, além de ausência de plaquetopenia e sintomas neurológicos. Afastadas demais etiologias de glomerulopatia e optado por desmame de corticoterapia.
A dosagem de toxina Shiga não foi possível por indisponibilidade no serviço bem como atividade de ADAMTS-13 e marcadores genéticos de SHU atípica.
Recebeu alta da Unidade de Terapia Intensiva após 5 dias, permanecendo em suporte dialítico na enfermaria com necessidade de sessões diárias para controle volêmico.
Foi encaminhada para serviço externo para realização de biópsia renal que confirmou o diagnóstico de Microangiopatia Trombótica, variante arteriolar. SHUa foi estabelecida como hipótese diagnóstica principal, sendo indicada introdução de eculizumabe. Contudo, uso da medicação foi impossibilitado pela indisponibilidade do medicamento no sistema de saúde público brasileiro.
6. DISCUSSÃO
A microangiopatia trombótica (MAT) é caracterizada por quadro de oclusão microvascular sistêmica ou intrarrenal que se apresenta laboratorialmente como anemia hemolítica microangiopática e trombocitopenia. (CAMPISTOL et al., 2015).
A paciente do caso apresentado teve diagnóstico confirmado de microangiopatia trombótica por biópsia renal e apresentava achados laboratoriais concordantes com o que está descrito na literatura para a síndrome, exceto pela contagem normal de plaquetas.
Entretanto, segundo Sallée et al., (2013), a trombocitopenia passou a ser questionada para a definição diagnóstica de Síndrome Hemolítico-Urêmica. Em estudo retrospectivo com 50 pacientes com diagnóstico histológico de MAT, foi demonstrado que 44% apresentaram contagem de plaquetas normal durante toda a evolução da doença. A maioria desses pacientes era do sexo feminino (70%) e a principal diferença entre os dois grupos foi a maior necessidade de diálise no grupo sem trombocitopenia (59 x 43%) (SERRES; ISENRING, 2018). Logo, o diagnóstico de MAT deve ser considerado em pacientes com anemia hemolítica, injúria renal e LDH aumentado, mesmo na ausência de trombocitopenia. (CAMPISTOL et al., 2015).
Na avaliação da síndrome, necessitamos prioritariamente investigar a possibilidade diagnóstica de PTT e STEC-SHU (CAMPISTOL et al., 2015).
Classicamente conhecida pelo envolvimento neurológico (BROCKLEBANK, et al, 2017), a PTT, embora, diagnóstico diferencial de MAT com alteração de função renal, tem associação importante com trombocitopenia persistente (< 30.000/mm³) e alterações de função renal discretas (BAGGA et al., 2019). Revisão de 2017 da função renal em 78 pacientes, em Oklahoma, nos Estados Unidos, mostrou que apenas 10% dos pacientes apresentavam lesão renal na admissão, sendo que 5% classificavam-se como KDIGO 3. No longo prazo, 6% evoluíram com Doença Renal Crônica e nenhum necessitou de diálise. (LITTLE et al., 2017).
Na suspeita diagnóstica de PTT, o uso do escore PLASMIC tem se mostrado bastante útil. Para a sua pontuação, utilizam-se critérios laboratoriais: Plaquetopenia < 30.000/mm³, sinais de hemólise (Reticulócitos > 2,5%, haptoglobina indetectável ou bilirrubina indireta > 2mg/dl), VCM < 90fL, INR < 1,5 e creatinina < 2,0mg/dl), além da avaliação da ausência de câncer em atividade ou de história de transplante de órgão sólido ou medula óssea. (BENDAPUDI et al., 2017).
Segundo Paydary et al. (2020), em metanálise avaliando 970 pacientes com hipótese de PTT, o escore com pontuação ≥ 6 apresentou 85% de sensibilidade e 89% de especificidade para atividade de ADAMTS-13 < 10%. Em coorte de 112 pacientes, com 21 diagnósticos confirmados, o valor preditivo negativo do escore foi de 98% (LI, et al. 2018).
No caso referido, a paciente apresentava PLASMIC escore de 3 e, portanto, baixo risco de PTT. A dosagem de ADAMTS-13 não estava disponível no serviço. Diante de escore PLASMIC baixo, somado à ausência de trombocitopenia e de alterações neurológicas, com a presença de lesão renal aguda grave, permitiu-se que a hipótese de PTT fosse excluída.
O protótipo da Síndrome Hemolítico-Urêmica é a STEC-SHU. Sua fisiopatologia está associada a uma lesão endotelial direta causada pela toxina Shiga. Clinicamente, está relacionada a quadro diarreico, que se inicia três dias após contaminação, além de dor abdominal, febre e vômitos. Inicialmente aquosa, a diarreia torna-se sanguinolenta cerca de três dias após os primeiros sintomas. As manifestações hematológicas e de disfunção renal costumam surgir após 10 dias do início do quadro (TARR; GORDON; CHANDLER, 2005). Embora exista forte relação entre diarreia e STEC-SHU, sinais de gastroenterite, como diarreia e dor abdominal podem aparecer em até um terço dos pacientes com SHU atípica. (LOIRAT et al., 2015).
O diagnóstico de STEC-SHU se dá pela demonstração da toxina Shiga em exame de coprocultura, podendo incluir cepas de E. coli O157:H7 e O104:H4 e Shigella ou então, pela detecção de genes da toxina Shiga nas fezes por PCR. (BAGGA et al., 2019). Tipicamente ocorre em crianças com idade inferior a 5 anos e apresenta uma incidência anual na Europa e América do Norte de 1,9-2,9 casos a cada 100.000 crianças nesta faixa etária. Em contrapartida, quando se considera a população com idade inferior a 15-18 anos, a incidência cai para 0,6-0,8 a cada 100.000 pessoas. Na América Latina, a incidência é dez vez maior que em outros continentes, chegando a 10-17 casos a cada 100.000 habitantes em crianças com menos de 5 anos (FAKHOURI et al., 2017).
No caso apresentado, a paciente não pertencia à faixa etária mais prevalente de acordo com os estudos e não teve diarreia sanguinolenta a despeito dos sinais de gastroenterite à entrada no serviço médico. Exame de coprocultura foi negativo. A pesquisa de Shiga toxina estava indisponível. Além disso, paciente trazia exames de serviço externo (realizados em 31/12/2022) que demonstravam urina I proliferativa com urinálise evidenciando proteinúria e hematúria, inferindo comprometimento renal inicial, contrariando a evolução típica dos casos de STECSHU.
A SHU atípica é uma doença rara e está relacionada a complicações e sequelas importantes secundárias a alterações da função renal. A lesão renal aguda grave acontece em 81% dos adultos e pode ser acompanhada por envolvimento glomerular, hematúria, proteinúria, edema e hipertensão. (MOTA, et al., 2021).
De acordo com Campistol et al. (2015), a SHU atípica é considerada uma doença ultrarrara. Dados disponíveis sobre incidência e prevalência são limitados, assim como a epidemiologia da doença. A incidência anual varia entre 1 a 2 casos a cada milhão de habitantes. Dados da Agência Médica Europeia, apontam prevalência de aproximadamente 3,3 pacientes a cada milhão de habitantes por ano entre pacientes abaixo dos 18 anos, com índices menores entre adultos. 60% dos casos se iniciam antes dos 18 anos, onde o acometimento não difere entre gêneros. Nos adultos, há predomínio do sexo feminino (LOIRAT et al., 2015). Outro estudo realizado na Europa com 16 milhões de pessoas mostrou prevalência de 4,96 por milhão de pessoas (WUHL, et al., 2014).
Quando se trata de fisiopatologia, a SHU atípica pode ser consequência da desregulação da via alternativa do complemento sobre superfícies celulares provocando dano endotelial e trombogênese (CAMPISTOL et al., 2015).
A fisiopatologia da SHU mediada por complemento está relacionada a uma hiperativação da via alternativa do complemento causada por autoanticorpos, como o anti-FH; mutações em genes reguladores do complemento, como Fator H, Fator I, MCP e trombomodulina ou nos genes componentes da C3-convertase, como fator B e C3. Em 60 a 70% dos pacientes, essas mutações ou autoanticorpos são identificáveis (LOIRAT et al., 2015).
Quando alguma dessas alterações está presente, a cascata da via alternativa sofre uma retroalimentação positiva de C3 e C3 convertase. A ligação da porção b do C3 (C3b) com a C3 convertase culmina em última instância na formação do complexo de ataque à membrana, o qual promove a lise endotelial. (MAC) (FERREIRA et al., 2019).
Figura 1. Descrição da via alternativa do complemento, apresentando genes e autoanticorpos envolvidos na SHUa e sua respectiva retroalimentação na cascata. Ação do eculizumabe na terapêutica da TMA associada ao complemento.
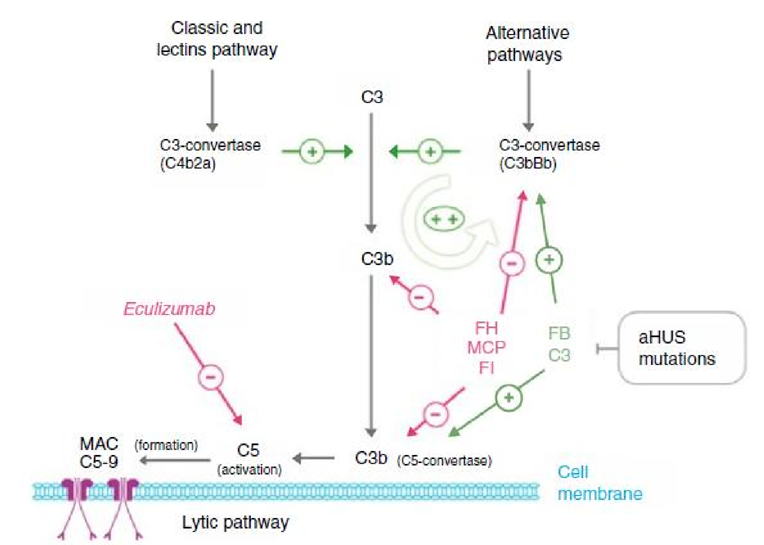
O aumento do conhecimento da patogênese da SHUa foi acompanhado pelo surgimento do Eculizumabe, um anticorpo IgG monoclonal que se liga ao fator C5 do complemento, impedindo a formação do complexo de ataque à membrana.
A decisão de usar terapia anticomplemento é baseada em diagnóstico presuntivo de SHU mediada por complemento, já que faltam testes confirmatórios imediatamente disponíveis. O uso de plasmaférese deve ser reservado para países subdesenvolvidos em que a terapia anticomplemento é limitada e o paciente apresenta recuperação insatisfatória. (RAINA et al., 2018).
Campistol et al. (2015) ainda afirma que para definir SHU mediada por complemento, outras causas secundárias devem ser excluídas.
A paciente foi admitida no setor de urgência/emergência com quadro de dispneia e hipertensão arterial secundária à hipervolemia, evidenciando-se posteriormente disfunção renal aguda grave em urgência dialítica. Além disso, trazia história de início dos sintomas há 20 dias da admissão com pródromo de diarreia autolimitada e vômitos, sendo caracterizado e conduzido como uma gastroenterite infecciosa aguda, além de urinálise de serviço externo com proteinúria e hematúria que fora tratada como infecção do trato urinário. Apresentava anemia normocítica normocrômica importante, porém sem plaquetopenia associada. Além disso, não possuía sinais ou sintomas prévios que pudessem caracterizar doença reumatológica autoimune. As investigações de colagenoses, demais culturas e sorologias da paciente vieram negativas, o que descartou a hipótese de SHU secundária.
O acometimento renal difere nas duas variantes principais da Síndrome Hemolítico-Urêmica. Segundo Polito; Kirsztajn (2010), a injúria renal aguda se apresenta em 55 a 70% dos casos, todavia é observada recuperação da função renal em mais de 70% desses pacientes. Nos 30% remanescentes, o prognóstico é desfavorável, apresentando doença renal crônica, dos quais apenas 1 a 4% permanecem com necessidade de terapia de substituição renal após terapia de suporte (FAKHOURI et al., 2017). De maneira oposta, na SHU mediada por complemento, a necessidade de terapia de substituição renal chega a 50% dos casos, com mortalidade atingindo até 25% desses pacientes (RAINA et al., 2018). Segundo Campistol et al. (2015), desde que o eculizumabe foi aprovado para o tratamento de SHU atípica em 2011, o prognóstico dos pacientes melhorou substancialmente, sendo aprovado como primeira linha de tratamento. Estudos mostraram aumento de plaquetas e melhora da função renal já na primeira dose(VAISBICH, et al., 2013). Há uma redução significante em mortalidade nos grupos que usaram tal anticorpo (RAINA et al., 2018). Eculizumabe obteve normalização hematológica em 90% dos pacientes e foi associado com um pequeno, porém significativo aumento na taxa de filtração glomerular. (FAKHOURI et al., 2017).
Quando o diagnóstico de SHUa é presumido, eculizumabe é recomendado como primeira linha de tratamento, no intuito de proteger a função orgânica (FRANCHINI, 2015). A despeito de ter seu maior benefício se iniciado nos primeiros 7 dias do início do quadro, como descrito por Walle, et al. (2016), é razoável considerar o tratamento mesmo em pacientes que encontram-se em terapia de substituição renal há 3 a 4 meses, devendo ser mantido o tratamento de 3 a 6 meses antes de concluir que não há benefício. A biópsia renal pode ser útil para definir a viabilidade de início ou manutenção da medicação, visto que essa apresenta benefícios em pacientes que apresentam lesões sugestivas de MAT, diferentemente do que é visto nos pacientes com biópsia demonstrando fibrose ou esclerose. (LOIRAT et al., 2015).
A paciente foi encaminhada para serviço externo de Nefrologia, onde realizou biópsia renal que identificou achados histológicos consistentes com Microangiopatia Trombótica, variante arteriolar. Foi indicada terapia com eculizumabe, porém, a indisponibilidade do medicamento no serviço público de saúde e os altos custos para sua aquisição, chegando a 300 mil dólares ao ano nos Estados Unidos conforme relatado por Raina et al., (2018) levou a equipe médica oferecer terapia de suporte. Paciente permaneceu em terapia renal substitutiva, sendo encaminhada para seguimento com equipe de transplante renal.
7. CONCLUSÃO
O diagnóstico de SHU mediada por complemento é um desafio por diversas razões. Trata-se de uma condição pouco prevalente e sem testes diagnósticos específicos. Sua confirmação exige diagnóstico diferencial com patologias muito semelhantes clinicamente e exames laboratoriais diagnósticos que estão majoritariamente indisponíveis na rede pública de saúde do nosso país. Mutações genéticas podem não ser encontradas em 30 a 40% dos casos e os resultados costumam demorar a ser disponibilizados.
A compreensão de como alcançar o diagnóstico precoce é fundamental, pois a redução da morbimortalidade dessa doença está relacionada a um início precoce de tratamento. No entanto, a medicação também é pouco disponível em nosso meio em decorrência do alto custo.
É necessária a valorização da doença por parte dos órgãos públicos, vislumbrando facilitar tanto o diagnóstico quanto o tratamento precoces, evitando desfechos não satisfatórios com elevada morbidade semelhante ao caso relatado.
REFERÊNCIAS
[1] MOAKE, Joel L.. Thrombotic Microangiopathies. New England Journal Of Medicine, v. 347, n. 8, p. 589-600, 2002. Disponível em: http://dx.doi.org/10.1056/nejmra020528. Acesso em 12 dez. 2022.
[2] THOMPSON, Gemma L.; KAVANAGH, David. Diagnosis and treatment of thrombotic microangiopathy. International Journal Of Laboratory Hematology, v. 44, n. 1, p. 101-113, set. 2022.Wiley. Disponível em: http://dx.doi.org/10.1111/ijlh.13954. Acesso em 12 dez. 2022.
[3] Polito MG, Kirsztajn GM. Thrombotic microangiopathies: thrombotic thrombocytopenic purpura / hemolytic uremic syndrome. Jornal Brasileiro de Nefrolologia, 2010 Jul-Sep;32(3):303-15. PMID: 21103695. Acesso em 12 dez. 2022.
[4] LOIRAT, Chantal et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatric Nephrology, v. 31, n. 1, p. 15-39, 11 abr. 2015. Springer Science and Business Media LLC. Disponível em: http://dx.doi.org/10.1007/s00467-015-3076-8. Acesso em 12 dez. 2022.
[5] JOSEPH, Catherine et al. Complement disorders and hemolytic uremic syndrome. Current Opinion In Pediatrics, v. 25, n. 2, p. 209-215, abr. 2013. Disponível em: http://dx.doi.org/10.1097/mop.0b013e32835df48a. Acesso em 22 jan. 2022.
[6] TONACO, Leandro C. et al. Púrpura trombocitopênica trombótica: o papel do fator von willebrand e da adamts13. Revista Brasileira de Hematologia e Hemoterapia, v. 32, n. 2, p. 155-161, 2010. Elsevier BV. Disponível em: http://dx.doi.org/10.1590/s1516-84842010005000043. Acesso em 18 dez. 2022.
[7] CAMPISTOL, Josep M. et al. Actualización en síndrome hemolítico urémico atípico: diagnóstico y tratamiento. Documento de consenso. Nefrología, v. 35, n. 5, p. 421-447, set. 2015. Elsevier BV. Disponível em: http://dx.doi.org/10.1016/j.nefro.2015.07.005. Acesso em 20 dez. 2022.
[8] GEORGE, James N.; NESTER, Carla M. Syndromes of Thrombotic Microangiopathy. New England Journal Of Medicine, v. 371, n. 7, p. 654-666, 2014. Disponível em: http://dx.doi.org/10.1056/nejmra1312353. Acesso em 22 dez. 2022.
[9] BAGGA, Arvind et al. Hemolytic uremic syndrome in a developing country: consensus guidelines. Pediatric Nephrology, v. 34, n. 8, p. 1465-1482, 2019. Springer Science and Business Media LLC. Disponível em: http://dx.doi.org/10.1007/s00467-019-04233-7. Acesso em 22 dez. 2022.
[10] FAKHOURI, Fadi et al. Haemolytic uraemic syndrome. The Lancet, v. 390, n. 10095, p. 681-696, 2017. Disponível em: http://dx.doi.org/10.1016/s01406736(17)30062-4. Acesso em 02 jan. 2023.
[11] VAISBICH, Maria Helena et al. Eculizumab for the treatment of atypical hemolytic uremic syndrome – Case report and revision of the literature. Jornal Brasileiro de Nefrologia, v. 35, n. 3, p. 237-241, 2013. FapUNIFESP (SciELO). Disponível em: http://dx.doi.org/10.5935/0101-2800.20130037. Acesso em 04 jan. 2023.
[12] FRANCHINI, Massimo. Atypical hemolytic uremic syndrome: from diagnosis to treatment. Clinical Chemistry And Laboratory Medicine, v. 53, n. 11, p. 16791688, 2015. Disponível em: http://dx.doi.org/10.1515/cclm-2015-0024. Acesso em 24 jan. 2023.
[13] WALLE, Johan Vande et al. Improved renal recovery in patients with atypical hemolytic uremic syndrome following rapid initiation of eculizumab treatment. Journal Of Nephrology, v. 30, n. 1, p. 127-134, 2016. Springer Science and Business Media LLC. Disponível em: http://dx.doi.org/10.1007/s40620-016-0288-3. Acesso em 04 jan. 2023.
[14] MITTAL, Kshitija et al. Challenges in management of atypical hemolytic uremic syndrome: bottle neck in resource limited settings. Transfusion Clinique Et Biologique, v. 29, n. 1, p. 98-100, 2022. Elsevier BV. Disponível em: http://dx.doi.org/10.1016/j.tracli.2021.07.002. Acesso em 07 jan. 2023.
[15] RAINA, Rupesh et al. Atypical Hemolytic-Uremic Syndrome: an update on pathophysiology, diagnosis, and treatment. Therapeutic Apheresis And Dialysis, v. 23, n. 1, p. 4-21, 29 out. 2018. Wiley. Disponível em: http://dx.doi.org/10.1111/1744-9987.12763. Acesso em 14 jan. 2023.
[16] SALLÉE, Marion et al. Thrombocytopenia is not mandatory to diagnose haemolytic and uremic syndrome. BMC Nephrology, v. 14, n. 1, 2013. Disponivel em: http://dx.doi.org/10.1186/1471-2369-14-3. Acesso em 28 jan. 2023.
[17] SERRES, S. A. de; ISENRING, P. Athrombocytopenic thrombotic microangiopathy, a condition that could be overlooked based on current diagnostic criteria. Nephrology Dialysis Transplantation, v. 24, n. 3, p. 1048-1050, 2008. Disponível em: http://dx.doi.org/10.1093/ndt/gfn687. Acesso em 30 jan. 2023.
[18] BROCKLEBANK, Vicky et. al. Thrombotic Microangiopathy and the Kidney. Clinical Journal Of The American Society Of Nephrology, v. 34, n. 8, p. 1465-1482, Springer Science and Business Media LLC. Disponível em: http://dx.doi.org/10.1007/s00467-019-04233-7. Acesso em 12 jan. 2023.
[19] LITTLE, Dustin J. et al. Long-term Kidney Outcomes in Patients With Acquired Thrombotic Thrombocytopenic Purpura. Kidney International Reports, v. 2, n. 6, p. 1088-1095, 2017. Disponível em: http://dx.doi/org/10.1016/j.ekir.2017 .06.007. Acesso em 14 jan. 2023.
[20] BENDAPUDI, Pavan K. et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. The Lancet Haematology, v. 4, n. 4, p. 157164, 2017. Disponível em: http://dx.doi.org/10.1016/s2352-3026(17)30026-1. Acesso em 17 jan. 2023.
[21] PAYDARY, Koosha et al. Diagnostic accuracy of the PLASMIC score in patients with suspected thrombotic thrombocytopenic purpura: a systematic review and meta-analysis. Transfusion, p. 1-11, 2020. Wiley. Disponível em: http://dx.doi.org/10.1111/trf.15954. Acesso em 22 jan. 2023.
[22] LI, A. et al. External validation of the PLASMIC score: a clinical prediction tool for thrombotic thrombocytopenic purpura diagnosis and treatment. Journal Of Thrombosis And Haemostasis, v. 16, n. 1, p. 164-169, 2018. Elsevier BV. Disponível em: http://dx.doi.org/10.1111/jth.13882. Acesso em 01 fev. 2023.
[23] TARR, Phillip I; A GORDON, Carrie; CHANDLER, Wayne L. Shiga-toxinproducing Escherichia coli and haemolytic uraemic syndrome. The Lancet, v. 365, n. 9464, p. 1073-1086, 2005. Disponível em: http://dx.doi.org/10.1016/s01406736(05)71144-2. Acesso em 24 jan. 2023.
[24] MOTA, Lennara Pereira et al. Atypical Hemolytic-Uremic Syndrome: Clinical manifestations and challenges in diagnosis. Research, Society And Development, [S.L.], v. 10, n. 9, p. 1-12, 2021. Disponível em: http://dx.doi.org/10.33448/rsd-v10i9.18365. Acesso em 31 jan. 2023.
[25] WUHL, E. et al. Renal replacement therapy for rare diseases affecting the kidney: an analysis of the era – EDTA Registry. Nephrology Dialysis Transplantation, v. 29, n. 4, p. 1-8, 2014. Disponível em: https://doi.org/10.1093/ndt/gfu030. Acesso em 31 jan. 2023.
[26] FERREIRA, Ana Claudia Goulart et al. Doenças associadas à deficiência do sistema complemento. Arquivos de Ciências da Saúde, v. 26, n. 1, p. 62, 2019. Faculdade de Medicina de São José do Rio Preto – FAMERP. Disponível em: http://dx.doi.org/10.17696/2318-3691.26.1.2019.1397.rg/10.1093/ndt/gfu030. Acesso em 31 jan. 2023.