PATHOPHYSIOLOGICAL SURVEY ON MCCUNE-ALBRIGHT SYNDROME
REGISTRO DOI: 10.5281/zenodo.10953255
Maria Luiza Sokacheski Meneghetti¹; Isabela Cristina Gonçalves da Silva1; Milena de França Martins¹; Yasmin Scolari Reco1; Maria Fernanda Piffer Tomasi Baldez da Silva².
Resumo
Introdução: A Síndrome McCune Albright, caracterizada pela tríade composta pela presença de displasia fibrosa, manchas café-com-leite e puberdade precoce, possui cunho genético a qual acomete o gene GNAS. Tal alteração acarreta uma substituição do aminoácido arginina por cisteína e histidina levando ao aumento das concentrações de AMPc gerando, assim, a hiperestimulação de diversos tecidos. Segundo Dumitrescu e Collins, é uma doença de baixa prevalência na população representada por 1/100.000 e 1/1.000.000 habitantes. Apesar desse fato e do entendimento de sua fisiopatologia, pouco se sabe a respeito dos fatores que levam a variabilidade fenotípica apresentada pelos portadores e o grau de acometimento nas determinadas regiões afetadas. Entre a repercussão clínica gerada, encontra-se a de natureza psíquica complementando a vasta gama de especialidades envolvidas no manejo e acompanhamento desses pacientes. Metodologia: Trata-se de um estudo de natureza exploratória, quanti-qualitativa, e observacional que se institui através de revisão de literatura e aplicação de questionário através das redes sociais aos pacientes portadores da síndrome, o qual objetivou estabelecer o predomínio, manifestações clínicas em comum e o grau de acometimento na qualidade de vida dos mesmos, a fim de entender a repercussão fisiopatológica da doença. Resultados: Observou-se um predomínio no sexo feminino, raça branca, com grande parte do diagnóstico até os 5 anos de idade, além da associação entre a quantidade de manchas café-com-leite e o aparecimento da displasia nos pacientes, o qual também apresentou predominio no quadro de dor semanal em menos da metade dos pacientes com um impacto significativo nas atividades diárias. No mais, foi observado que, apesar da ampla gama de especialidades abordadas no manejo e no tratamento das repercussões da síndrome, ainda se encontra insatisfação dos participantes sobre as informações disponíveis a respeito da mesma. Discussão: Existe o predomínio de características e concordância com alguns dados obtidos através da revisão sistemática da literatura, porém também se obteve divergência em pontos específicos os quais mostraram que a tendência natural da doença se alterou, como exemplo maior incidência na raça branca. No mais, a associação entre a quantidade de manchas café-com-leite e o aparecimento da displasia fibrosa foi ponto a ser levado em consideração. Considerações finais: Conclui-se que, após a análise exploratória e observacional, a Síndrome McCune Albright é uma doença a ser entendida com uma abordagem multidisciplinar a fim de suprir as variadas necessidades individuais de seus portadores. Além disso, sugere-se a necessidade de mais estudos que abordem a relação entre o número de manchas café-com-leite e o desenvolvimento da displasia fibrosa.
1 INTRODUÇÃO
A síndrome de McCune Albright (SMA) configura-se como uma alteração genética rara com prevalência estimada que varia entre 1/100.000 e 1/1.000.000 ocorrendo igualmente nas diferente raças, porém com uma prevalência maior no sexo feminino em relação a puberdade precoce (DUMITRESCU e COLLINS, 2008). Sua incidência é desconhecida, tendo alterações fisiológicas que variam de indivíduo para indivíduo.
Originalmente a síndrome, descrita em 1936, como uma tríade fibrosa óssea, máculas cutâneas café com leite e puberdade precoce. No entanto, com o tempo foi possível observar outras endocrinopatias, como hipertireoidismo, cistos ovarianos produtores de estrogênio, excesso de hormônio do crescimento, perda de fosfato renal com ou sem raquitismo/osteomalácia e síndrome de Cushing podem ser observadas em associação com a tríade original (GARCIA NETO, et al. 2021).
A fisiopatologia de SMA se estabelece por uma mutação que ocorre ativando o gene GNAS que acarreta em uma substituição do aminoácido arginina por cisteína e histidina, aumentando as concentrações de AMPc intracelular, levando a uma hiperfunção de vários tecidos (SALLUM, et al. 2007).
Outro viés importante para ser ressaltado, é a displasia fibrosa óssea como um desafio devido às múltiplas doenças em que pode estar associada, gerando confusão diagnóstica e de proposta terapêutica (DUMITRESCU e COLLINS,2008). É de suma importância que a displasia quando identificada seja correlacionada com sua patologia base, uma vez que influenciam na conduta terapêutica. Em relação à Síndrome de McCune Albright a padronização dos achados clínicos é ainda mais complicada devido à enorme variedade de gravidade e apresentações da displasia nos diferentes portadores. Ainda assim, outras problemáticas podem ocorrer, como, comprometimento neurológico e incapacidade devido a deformidades dos ossos, por isso faz necessário que se tenha um maior conhecimento das características sindrômicos por parte dos profissionais de saúde, que só será possível com o agrupamento de dados e divulgação de novas pesquisas acerca do assunto.
A síndrome apesar de ter uma tríade sintomatológica bem definida, o quadro clínico de cada paciente é muito variável. Apesar disso os relatos de caso e o agrupamento desses sinais e sintomas muitas vezes não acontece de forma satisfatória a fim de enriquecer a base de dados sobre a síndrome e promover discussões acerca do assunto e possíveis melhorias nos futuros diagnósticos e tratamentos.
Por ser considerada rara, a síndrome ainda se encontra uma dificuldade em relação ao conhecimento e abordagem médica frente a mesma, uma vez que, existem muitos pontos a serem observados, em diferentes especialidades, por meio de exames laboratoriais e de imagem para o correto diagnóstico. Frente a isso, o pouco conhecimento médico leva a um atraso no diagnóstico da doença apesar tendência natural da doença ao aparecimento precoce de componentes da tríade. Assim, o Sistema Único de Saúde (SUS), apresenta falhas nesse processo, levando ao atraso do reconhecimento desta patologia (DUMITRESCU e COLLINS,2008).
Desse modo, identificamos a necessidade de fomentar o debate científico, ampliando a gama de informações acerca da síndrome de McCune-Albright, sua fisiopatologia e manifestações clínicas. Com este fim, buscamos obter informações diretas dos portadores da síndrome com o intuito de facilitar o diagnóstico para a classe médica e propiciar informações mais claras para a população afetada.
Visto isso, o trabalho em questão visa reunir as informações concretas da síndrome a partir de obras já publicadas anteriormente a fim promover um parecer mais claro a respeito de sua fisiopatologia e manifestações clínicas. Somando a isso, a segunda etapa consiste em obter resultados atuais dos portadores com o intuito de facilitar o diagnóstico e fornecer informações chave para o conhecimento da mesma.
2 FUNDAMENTAÇÃO TEÓRICA
Descrita pela primeira vez na história por McCune e Albright em 1936 e 1937, respectivamente, foi conceituado a tríade de displasia fibrosa polióstica, que culmina em claudicação, dor ou fraturas e máculas café com leite geralmente em um hemicorpo. Estes sinais aparecem inicialmente, mas não necessariamente em ordem, pois a doença não tem uma cronologia pré-estabelecida. É comum a presença de endocrinopatias hiperfuncionantes como a puberdade precoce, hipertireoidismo, hipercortisolismo, raquitismo hipofosfatêmico e hipersomatotropismo.
É de suma importância compreendermos que a etiologia está atrelada a fatores genéticos, embriológicos e biopsicossociais. A base molecular da SMA é uma mutação ativadora no gene GNAS1, que codifica uma subunidade da proteína G. Isto resulta na ativação constitutiva das proteínas Gαs, que estimulam autonomamente o sistema adenilil ciclase, resultando em declínio funcional dos tecidos afetados. (FARHAT et al., 1999). Além disso, a mutação no gene interfere na linhagem osteoblástica o qual ocasiona a displasia fibrosa encontrada na síndrome por consequente alteração na produção osteogênica nos ossos, incluindo os da medula óssea (ATALLA, A. et al.).
Do ponto de vista embriológico, pela síndrome apresentar uma clínica extremamente variável e não haver registros de hereditariedade, sugere-se que seja uma mutação do tipo somática ou pós zigótica, presumindo-se que esta alteração genética tenha ocorrido antes da formação do disco trilaminar pois os tecidos afetados possuem origem tanto ectodérmica, endodérmica como mesodérmica. (RINGEL; SCHWINDINGER; LEVINE, 1996). Contrapondo-se a isso, segundo Candida, M. a transmissão paterna e materna de alelos mutados levam a repercussões clínicas de acordo com o alelo herdado, como no caso de osteodistrofia hereditária de Albright de herança paterna e o pseudohiparatiroidismo de herança materna. (CANDIDA, M. 2002).
Em relação às manchas café com leite se caracterizam como aumento da produção de melanina sem alteração da quantidade de melanócitos. Diante disso, as máculas se expressam como um sinal clínico precoce e sugestivo que pode auxiliar em um diagnóstico mais rápido da doença, uma vez que, tendem a aparecer ainda no período neonato e antes do 1 ano de vida (GARCIA NETO V, 2021).
Compondo a tríade que caracteriza a síndrome, a puberdade precoce se caracteriza por amadurecimento precoce do eixo hipotálamo-hipófise-gonadal (HHG) que ocorre antes dos 8 anos de idade em meninas e antes dos 9 anos em meninos. Na doença, segundo Grumbach. MM, a puberdade precoce se estabelece como uma pseudopuberdade a qual é independente do hormônio central, GnRH, onde a estimulação do eixo ocorre perifericamente (NEVES DE CARVALHO, et al. 2007).
No aspecto biopsicossocial constatou-se por meio de relatos de caso que os pacientes portadores da síndrome possuem geralmente problemas psicológicos de aceitação social devido às deformidades ósseas, sobretudo quando ocorre assimetria facial (FARHAT et al., 1999), diante deste contexto é relevante destacar que boa parte dos pacientes iniciam o quadro displásico em idade escolar, o que pode ser um fator determinante para a ocorrência de bullying, aparecimento de transtornos depressivos e até evasão escolar. Diante do exposto, é possível compreender a necessidade de uma alternativa terapêutica que possa controlar a progressão da doença óssea, melhorando assim a qualidade de vida destes pacientes, pois as terapêuticas atualmente disponíveis apenas se limitam aos sintomas e não ao cerne da patologia.
O diagnóstico atualmente limita-se à identificação da tríade clássica que consiste na displasia fibrosa óssea, manchas café com leite visíveis pela derme e na disfunção endócrina, sendo mais comum o distúrbio do crescimento chamado de puberdade precoce. É de praxe a realização de exames complementares, estes que não são obrigatórios para a comprovação da síndrome mas que auxiliam grandemente em sua caracterização pois temos conhecimento do caráter multifacetado desta patologia. Estes exames são principalmente a imagem por tomografia computadorizada da região acometida pela displasia para avaliação do grau de invasão óssea e testes hormonais séricos endocrinológicos para avaliar a disfunção do eixo.
Vale ressaltar que uma abordagem multidisciplinar e baseada na integralidade é fundamental para compreender todas as demandas individuais que cada paciente afetado por esta síndrome possui. Sendo assim, o tratamento, apesar de não curativo, é singular para cada pessoa e deve abordar cada uma das manifestações, desde cirurgias corretivas de deformidades para os casos de displasia fibrosa óssea que necessitem, até fármacos que irão suprimir a produção hormonal sexual.
Independente do caso clínico de cada paciente, é um pilar do tratamento o acompanhamento longitudinal de cada manifestação para monitoramento, registro e adequação da conduta de acordo com a progressão da doença.
3 METODOLOGIA
No presente estudo foi utilizada uma abordagem mista, ou seja, tanto quantitativa quanto qualitativa, consistindo em uma pesquisa de natureza básica exploratória que busca discorrer sobre a Síndrome de McCune Albright por meio de fundamentações teóricas revisadas, ampliando o conhecimento científico acerca do objeto de estudo. A primeira etapa do projeto se faz através de um estudo de pesquisa bibliográfica, a amostragem corresponde à referenciais teóricos, como artigos, dissertações e teses, os quais foram retirados de bases de dados e revistas científicas. Dentre as bases de dados utilizadas estão o PubMed, LILACS, Google Acadêmico, Scielo (Scientific Eletronic Library Online) e Villar.
Os materiais selecionados foram os publicados desde 1999, usando como base as seguintes palavras-chave: Síndrome de McCune Albright ; Displasia óssea; Puberdade precoce; Endocrinopatias hiperfuncionantes
Com a finalidade de realizar um estudo detalhado e com informações verídicas, mais de 10 materiais publicados foram analisados e estudados, buscando sempre utilizar referências indexadas e publicadas ao longo de 25 anos em relação ao tema abordado. A análise dessas fontes foi realizada de modo a descrever seus conceitos e relações com a síndrome.
A segunda etapa foi a aplicação de um questionário por meio de um formulário online através das redes sociais aos portadores com ênfase na idade de diagnóstico; sexo; sinais, sintomas, local e o nível de fraturas, conhecimento sobre a evolução clínica da síndrome, além de tratamento. Como critérios de inclusão fizeram parte da pesquisa todos os membros de grupos de redes sociais que apresentem a síndrome e que desejaram participar da pesquisa, mediante a assinatura Termo de Esclarecimento Livre e Esclarecido, para maiores de 18 anos, ou Termo de Assentimento Livre e Esclarecido, para pacientes entre 12 e 18 anos, acompanhado do Termo assinado pelos responsáveis.
Os dados foram armazenados em planilhas do Programa Excel e analisados para indicar a frequência e incidência do objeto de estudo. Os resultados obtidos são apresentados em tabelas descritivas que foram relacionadas e apresentadas.
4 RESULTADOS
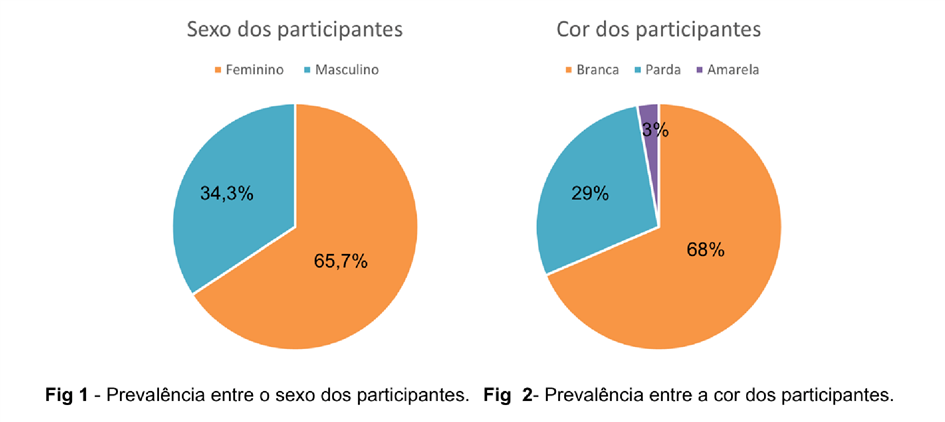
Em relação aos dados epidemiológicos encontrados (Figura 01), foi constatado uma prevalência da Síndrome no sexo feminino, aproximadamente 65,71% em comparação ao sexo masculino, 34,28%. Em relação à raça, observou-se que cerca de 68% dos indivíduos que responderam o questionário eram brancos, 28% são pardos e 2,8% amarelos (Figura 02).
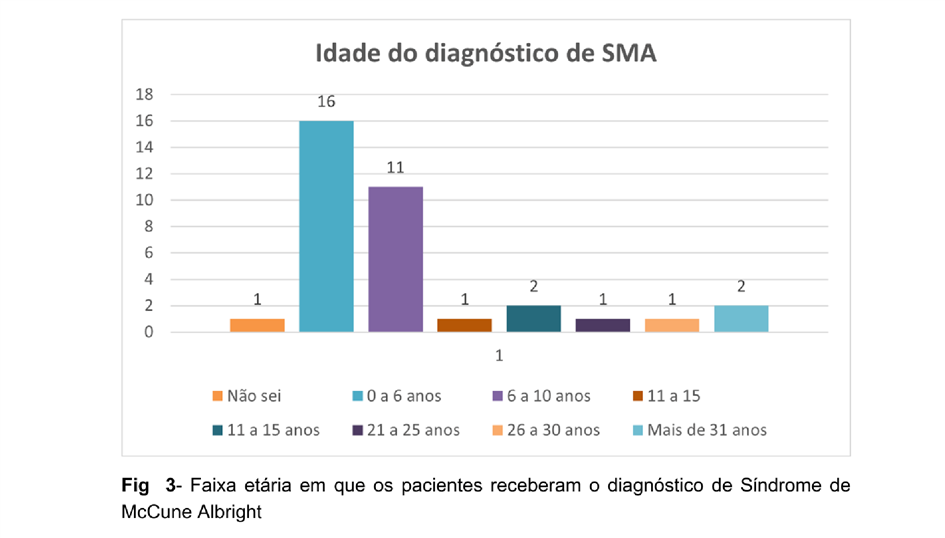
Sobre a idade do diagnóstico, foi constatado que cerca de 44,44% dos pacientes receberam o diagnóstico até os 5 anos de idade, 30,55% entre os 6 e 10 anos e 19,44% obtiveram o mesmo durante o período da adolescência e fase adulta. (Figura 03).
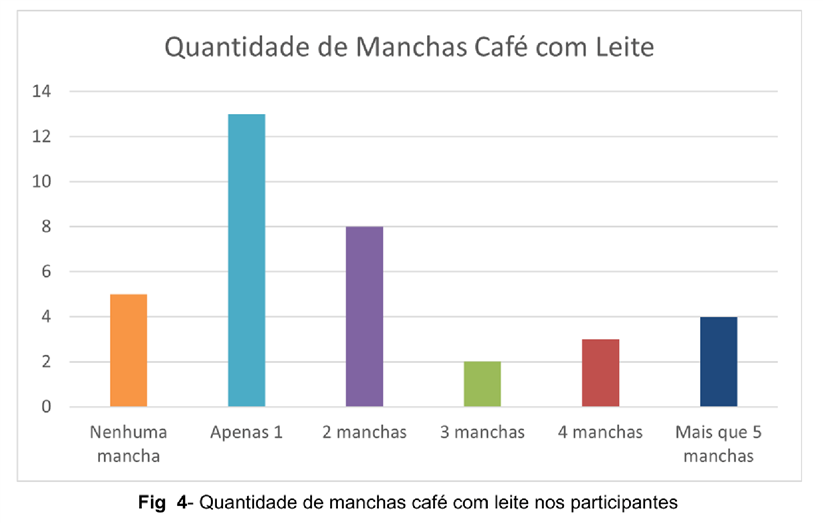
No que diz respeito às manchas café com leite, foi observado que cerca de 94,11% apresentam uma associação entre a quantidade de manchas café com leite e o desenvolvimento da displasia óssea, onde, para haver essa associação, o número de manchas prevalentemente deve ser maior ou igual a 2. Nessa análise, 17 pacientes apresentam 2 ou mais manchas , 13 possuem apenas 1 mancha e 5 não apresentam nenhuma. Complementado isso, 83,33% daqueles que responderam que não apresentam alteração/deformidade óssea decorrente da displasia, possuem apenas de 0 a 1 mancha.
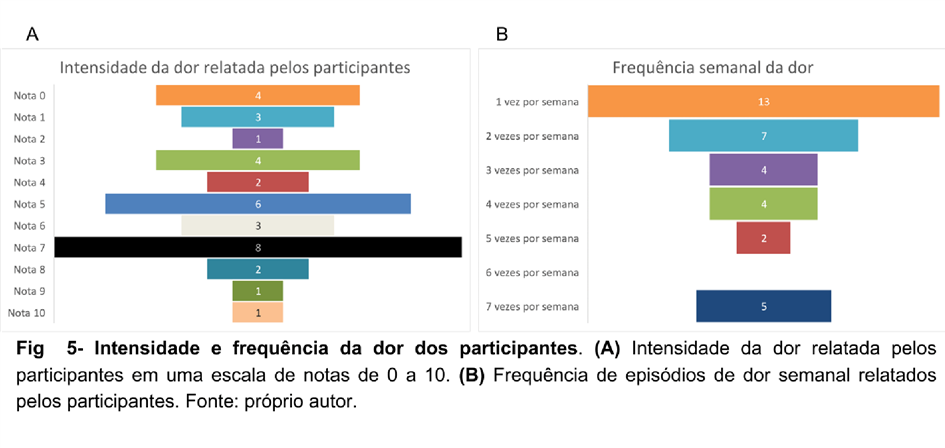
1,42% dos pacientes informaram que não sentem dor atualmente, sendo esta nota 0 em uma escala de dor de 0 a 10, como observado na Figura 05-A. Nessa análise, é possível observar também a frequência semanal de dor dos participantes, que têm predominância de 37,14% das vezes, aparecendo 1 episódio por semana. Não foi possível estabelecer uma relação entre a frequência e intensidade dos episódios dolorosos causados pela displasia, assim, observa-se a necessidade de uma avaliação individualizada frente a displasia.
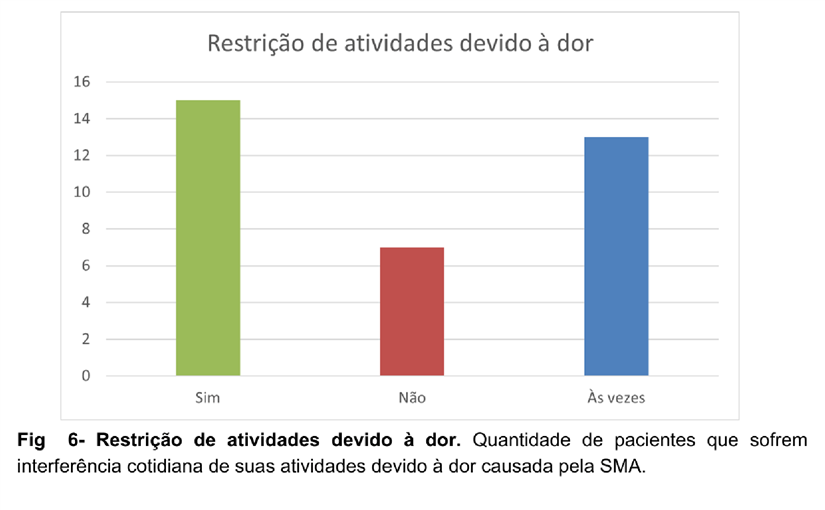
Em relação ao impacto na qualidade de vida dos portadores da SMA, foi visto que 44,44% dos pacientes frequentemente abandonam suas atividades por conta da dor, 19,44% não sofrem com sua influência e 36,11% sofrem ocasionalmente com essa questão. (Figura 6).
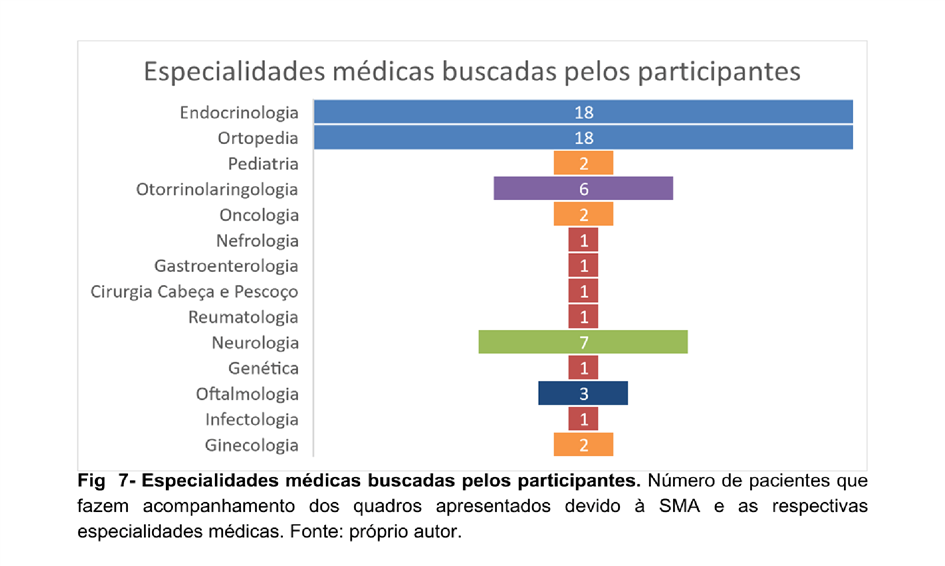
Desse modo, diversas especialidades foram citadas nesta pesquisa, porque desempenham papéis significativos no manejo dessa condição complexa. Dentre as mais procuradas pelos pacientes estão: Endocrinologia, Ortopedia, Neurologia e Otorrinolaringologia, respectivamente (Figura 07). Essa abordagem interdisciplinar é essencial, pois a SMA afeta não apenas o sistema esquelético, mas frequentemente vem a apresentar implicações em outros sistemas do corpo como o ginecológico, neurológico, oftálmico e do aparelho auditivo.
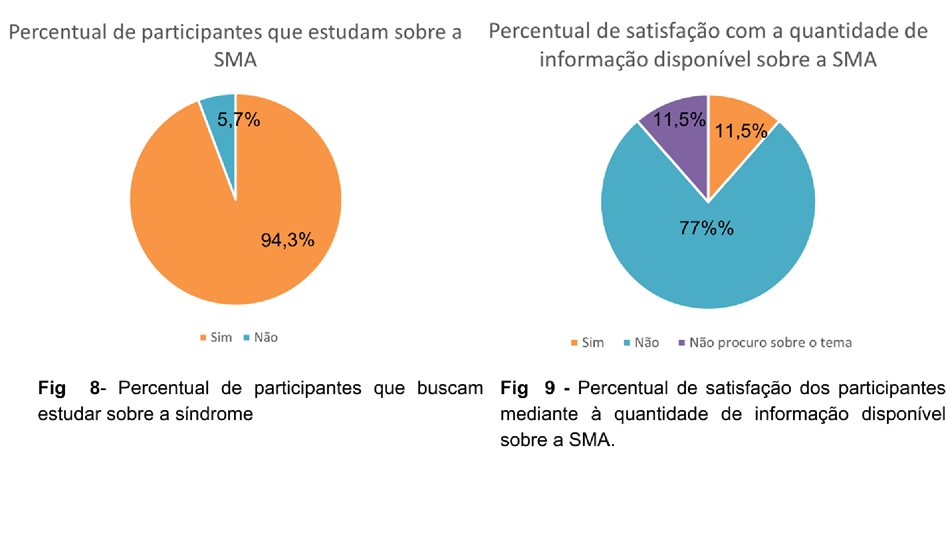
Com base na atitude dos pacientes em relação ao conhecimento médico sobre a displasia óssea, observou-se uma tendência positiva, uma vez que, a maioria dos pacientes
(94,3%) demonstrou interesse em buscar informações adicionais sobre sua condição (Figura 08), indicando um desejo de se envolver ativamente em seu próprio cuidado. No entanto, as informações que estão disponíveis na internet e base de dados não são totalmente esclarecedoras, sendo que 77% dos pacientes não acham satisfatória a qualidade de informações encontradas (Figura 09). Dessa forma, foi observado uma relevância estatística em relação à insatisfação dos pacientes com o conhecimento médico apresentado sobre a SMA, uma vez que, 51,4% demonstraram não estarem satisfeitos com as informações fornecidas pelos seus respectivos profissionais da saúde.
5 DISCUSSÃO
A prevalência maior no sexo feminino foi constada e mantida segundo Dumitrescu e Collins, contraponto, no entanto, o que foi defendido pelo mesmo uma vez que o resultado da pesquisa demonstrou uma incidência maior da síndrome na raça branca (DUMITRESCU e COLLINS, 2008). Em relação ao diagnóstico, grande maioria dos pacientes o recebeu entre a entre a primeira e segunda infância o qual estabelece um bom prognóstico, visto que intervenções médicas podem ser feitas tendo uma eficácia maior do tratamento. Já o aparecimento das manchas café-com-leite na primeira infância é uma característica da doença a ser levado em consideração uma vez que é um preditivo da síndrome. Nessa análise, foi possível observar a relação existente entre o número (2 ou mais) manchas café-com-leite e o desenvolvimento da displasia fibrosa. Tal fato também corrobora para uma intervenção médica precoce visando minimizar danos maiores futuros, visto que, além de as máculas surgirem precocemente, a faixa etária entre 0 e 10 é crucial para o desenvolvimento ósseo e é o período de maior remodelamento do mesmo visto que as epífises ósseas ainda não se fecharam. Apesar do levantamento trazido pela pesquisa, essa associação entre a displasia e o número de máculas nos pacientes necessita de uma abordagem mais específica. Concomitantemente, é possível fazer uma relação entre a idade de aparecimento dos sintomas com a gravidade do quadro, sendo que nos casos considerados mais graves a SMA pôde ser reconhecida logo após o nascimento e nos menos graves em qualquer fase da infância (SALLUM, 2008). Ponto este, a qual um diagnóstico nessa fase pode influenciar positivamente o curso da condição.
Outro ponto importante é que não se fez possível a obtenção de uma relação entre frequência e intensidade, mesmo a incidência de dores ósseas acometer menos da metade dos entrevistados e de forma semanal. No entanto, tal fato não corrobora para uma menor repercussão na vida dos pacientes, uma vez que, mais de 40% dos participantes relatam abandono de suas atividades diárias devido aos sintomas apresentados. Tal fato reforça ainda mais a individualidade das manifestações clínicas. Frente a isso, diversas especialidades médicas fazem parte da procura por parte dos pacientes para o tratamento das diversas repercussões clínicas que a síndrome apresenta, entre elas, a área endocrinológica e ortopédica lideram esse ranking, visto que a disponibilização de conteúdo a respeito da doença, assim como o baixo conhecimento médico geral sobre a mesma deixa a desejar, requisitando ainda mais a busca por médicos especializados.
6 CONCLUSÃO/CONSIDERAÇÕES FINAIS
Com base nas informações elencadas no decorrer deste artigo pode-se concluir que os resultados sugerem a necessidade de uma abordagem multiprofissional a estes pacientes, pois as manifestações, apesar de possível semelhança entre os casos, são totalmente individuais e a síndrome parece não ter um curso natural bem estabelecido.
Sendo assim, os pacientes devem ser seguidos por pediatras, endocrinologistas, ortopedistas, psicólogos, assistentes sociais e o constante apoio da família para terem uma melhor qualidade de vida. Por fim, ressalta-se a necessidade de pesquisas mais direcionadas nas duas características que compõem a síndrome (número de máculas e o desenvolvimento da displasia) a fim de entender os mecanismos que levam a mesma para evitar ou minimizar danos futuros.
REFERÊNCIAS
- ATALLA, A. et al. Displasia fibrosa: relato de caso e revisão de literatura. Rev Med Minas Gerais, v. 20, n. Esp, p. 399–403, [s.d.].
- CANDIDA, M.; VILLARES FRAGOSO, B. Manifestações Endócrinas das Mutações da Proteína Gsα e do Imprinting do Gene GNAS1. Disponível em: . Acesso em: 28 maio. 2022. Disponível em: . Acesso em: 30 maio. 2022.
- COLLINS, M. T.; SINGER, F. R.; EUGSTER, E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet Journal of Rare Diseases, v. 7, n. Suppl 1, p. S4, 2012.
- DE CASTRO, L. F. et al. Activation of RANK/RANKL/OPG Pathway Is Involved in the Pathophysiology of Fibrous Dysplasia and Associated With Disease Burden. Journal of Bone and Mineral Research, v. 34, n. 2, p. 290–294, 29 nov. 2018.
- DUMITRESCU, C. E.; COLLINS, M. T. McCune-Albright syndrome. Orphanet Journal of Rare Diseases, v. 3, n. 1, 19 maio 2008.
- FARHAT, H. L. et al. Evolução da displasia fibrosa óssea na síndrome de McCune Albright. Arquivos brasileiros de endocrinologia e metabologia, v. 43, n. 5, p. 351–359, 1999.
- GARCIA NETO, V. et al. McCune-Albright syndrome – A case report with transmission electron microscopy. Anais Brasileiros de Dermatologia, v. 97, n. 1, p. 58–62, jan. 2022.
- CARVALHO. N. M. et al. Puberdade precoce: a experiência de um ambulatório de ginecologia infanto juvenil. Rev. Bras. Ginecol. Obstet. 29 (2), fev. 2007.
- VILAR, Lucio. Endocrinologia clínica, 7° edição. Guanabara Koogan, 2022.
- SALLUM, A. C. R. et al. Hipertireoidismo relacionado à síndrome de McCune Albright: relato de dois casos e revisão da literatura. Arquivos brasileiros de endocrinologia e metabologia, v. 52, n. 3, p. 556–561, 2008.
- PERES, H. C. R. et al. Hipertireoidismo associado à síndrome de McCune Albright: Relato de caso/ Hyperthyroidism associated with McCune Albright syndrome: a case report. Brazilian Journal of Health Review, v. 4, n. 6, p. 24007–24014, 2021.
- RINGEL, M. D.; SCHWINDINGER, W. F.; LEVINE, M. A. Clinical Implications of Genetic Defects in G Proteins: The Molecular Basis of McCune-Albright Syndrome and Albright Hereditary Osteodystrophy. Medicine, v. 75, n. 4, p. 171–184, jul. 1996.
¹Discente do Curso Superior de Medicina da Universidade Cesumar Campus Maringá e-mail: marialuizameneghetti@hotmail.com; abela_cristina@hotmail.com ; milenafrancamartins@gmail.com ; yasscolari@icloud.com
²Docente do Curso Superior de Medicina da Universidade Cesumar Campus Maringá. Mestre em Genética pela UEM, Doutora em Genética pela UFPE. e-mail: maria.baldez@docentes.unicesumar.edu.br