REGISTRO DOI: 10.5281/zenodo.7817940
Deane Pereira Furtado Guedes1
Cleide Roberta de Arruda Franco1
Eduardo Faustino Coelho Sousa1
Evely Lara Teixeira Melo1
Flávio Gabriel Rosas Costa1
Leonardo Machado Alecrim1
Viviane da Conceição Gomes Siqueira1
Ydrielly Veras Teles2
RESUMO: Descrita como um erro metabólico hereditário autossômico recessivo, a Glicogenólise Tipo I ou Doença de Von Gierke, compreende uma deficiência da enzima glicose 6-fosfatase, responsável pela reação final da glicogenólise e gliconeogênese. Esta revisão bibliográfica trata da temática atual da doença de Von Gierke, tendo como objetivo a breve descrição e resumo do que foi achado nos últimos anos relacionando a fisiopatologia, diagnóstico, prognóstico e tratamento. A busca sistemática de ensaios clínicos randomizados e não randomizados foi realizada por meio das bases eletrônicas de dados utilizando-se o descritor Glicogenose do Tipo Ia em inglês, sendo os resultados tratados como objeto de análise. Nota-se que os pacientes com essa patologia não convertem a glicose 6-fosfato em glicose livre para a corrente sanguínea, resultando em um acúmulo excessivo de glicogênio e gordura no fígado, rins e intestinos. Dentre os 13 tipos de glicogenoses, a tipo Ia é a mais comum e uma das mais graves. Indivíduos com GSD Tipo Ia apresentam hipoglicemia, hipertrigliceridemia, hiperuricemia, hepatomegalia e em alguns casos nefromegalia. Conhecer os aspectos bioquímicos da doença é de fundamental importância para que se desenvolva um manejo adequado no tratamento e assim, minimizar os seus efeitos deletérios.
PALAVRAS-CHAVE: Glicogenose; Glicose 6-fosfatase; Deficiência enzimática.
INTRODUÇÃO
Há um consenso entre os aurores que a GSD1a é uma doença de cunho metabólico hereditário recessivo, na qual existe uma deficiência na enzima glicose 6-fosfatase que impede a conversão do glicogênio em glicose livre e fosfato inorgânico para o sangue. Entender as vias metabólicas da glicose é extremamente importante porque algumas células utilizam quase exclusivamente a glicose como fonte de energia.
O organismo possui uma reserva de glicose em forma de glicogênio, que são armazenados nos músculos, fígado, rins e intestinos. Nos três últimos, é necessário a ação catalítica da enzima glicose 6-fosfatase para que se converta o glicogênio em glicose quando necessário através de vias normais de glicogenólise, sendo crucial para a manutenção dos níveis de glicose sanguínea em estado de jejum.
Como sintomas iniciais da Doença de Von Gierke ocorrem logo em seguida ao nascimento devido à hipoglicemia e episódios não responsivos à administração de glucagon. Os principais sintomas são tremores, irritabilidade, hiperventilação, cianose, apneia, convulsões, palidez, suor, edema, disfunção cerebral, coma e até morte.
Através da seguinte revisão de literatura objetiva a breve descrição e resumo do que foi achado nos últimos anos, na mesma linha de pensamento com relação à fisiopatologia, diagnóstico, prognóstico e tratamento.
METODOLOGIA
A busca sistemática de ensaios clínicos randomizados e não randomizados foram realizada por meio das seguintes bases eletrônicas de dados PUBMED (https://pubmed.ncbi.nlm.nih.gov/) e SCIELO, foi utilizado o descritor Glicogenose do Tipo Ia em inglês (Glycogen Strage Disease type 1A) como elementos de busca. Utilizou-se como critérios de exclusão, artigos com mais de 5 anos e não-gratuitos, modelos experimentais com animais e que abordassem terapias gênicas, obtendo-se 17 artigos achados.
Os artigos da pesquisa foram tratados como objeto de análise do assunto Glicogenose do Tipo IA, sendo conduzida em março de 2023. Primeiramente, os títulos e resumos de todos os artigos identificados pela estratégia de busca foram avaliados independentemente e em duplicata por dois avaliadores. Todos os resumos que não forneciam informações suficientes sobre os critérios de inclusão e exclusão foram selecionados para leitura. Na segunda etapa, os mesmos revisores avaliaram artigos completos, independentemente e em duplicata, para selecionar aqueles em conformidade com os critérios de elegibilidade.
Visando assegurar a veracidade dos dados expostos neste trabalho, afirmo que todos os dados foram subjetivados conforme dados já existentes e revisados. As diferenças entre os revisores foram resolvidas por consenso entre os pesquisadores.
RESULTADOS E DISCUSSÃO
Este artigo de revisão bibliográfica tem como escopo abordar os aspectos bioquímicos e clínicos da doença de armazenamento de glicogênio tipo Ia (GSD tipo Ia), que configura a deficiência do complexo da enzima glicose 6-fosfatase, que é responsável pela dissociação da glicose e o fosfato.
De acordo com o portfólio bibliográfico, os artigos compreenderam as publicações dos últimos cinco anos. Os artigos estão agrupados por ano na tabela 1

Tabela 1 – portfólio bibliográfico incluído no estudo
FISIOPATOLOGIA
A GSD tipo I é a mais comum e uma das mais graves, com mais de 80% dos casos atribuídos a uma deficiência na enzima glicose 6-fosfatase (G6Pase α; Tipo Ia) (ELLINGWOOD & CHENG, 2018). Segundo Lima & Beal, 2011 (apud CORI; CORI, 1952), a deficiência da glicose 6-fosfatase na DGA-I foi demonstrada pela primeira vez em 1952 por Cori e Cori, e na primeira patologia específica a enzima foi identificada como uma desordem hereditária. Em 1929, Von Gierke descreveu as características dessa doença em um paciente.
O gene responsável pela doença, G6PC, foi mapeado para o cromossomo 17q21, e mais de 100 mutações no G6PC foram relatadas até agora. Embora esteja presente em todas as etnias, algumas mutações são mais prevalentes em certas etnias. As mutações de R83C e Q347X são frequentes na população judaica asquenaze (Niba et al., 2021). De acordo com Chaves (2009), a glicogenose tipo I representa 25% do total das glicogenoses e os indivíduos com esta doença são incapazes de converter a glicose 6-fosfato em glicose livre para a circulação sanguínea. Dessa forma se acumula glicogênio em excesso no fígado, rins e mucosa intestinal.
Olgac et al. (2021) pontuam que devido à atividade deficiente desta enzima, um acúmulo excessivo de glicogênio ocorre no fígado, rim, músculos esqueléticos e mucosa intestinal. Como a homeostase da glicose não pode ser mantida durante o jejum, episódios de hipoglicemia com risco de vida podem ser observados.O acúmulo de glicogênio em hepatócitos e túbulos renais proximais leva à hepatomegalia e nefromegalia (Sol et al., 2022).
ASPECTOS BIOQUÍMICOS
Ellingwood & Cheng (2018), afirmam que em humanos o glicogênio é a principal forma de armazenamento da glicose e o principal meio de descarte de glicose não oxidativa nos tecidos muscular e hepático. Durante momentos de necessidade, o glicogênio é rapidamente decomposto para produzir glicose.
No fígado, a quebra do glicogênio durante as condições de jejum contribui para a produção hepática de glicose, que é crucial para manter os níveis de glicose no sangue, para apoiar as necessidades de outros tecidos (Figura 1) (ELLINGWOOD & CHENG, 2018).
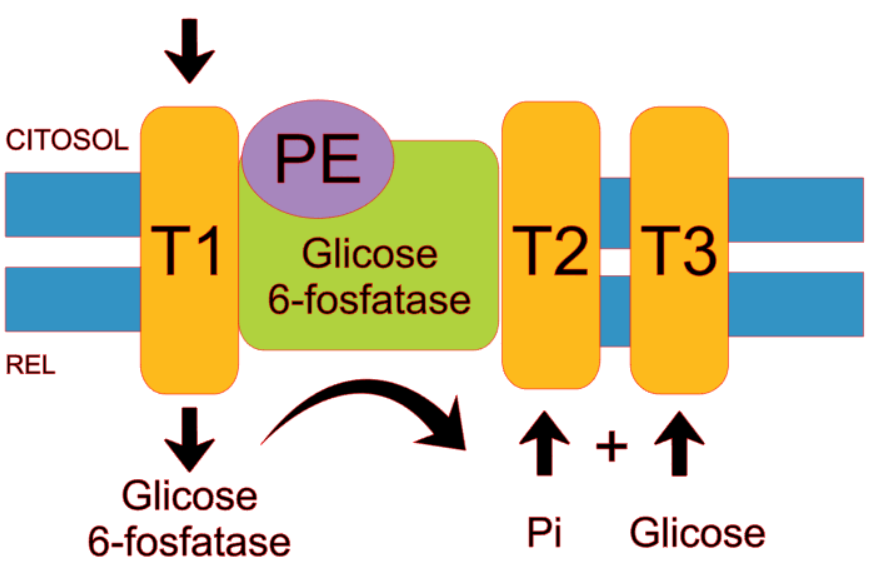
Figura 1 – Representação esquemática da dissociação do fosfato da glicose mediada pelo complexo enzimático da glicose 6-fosfatase (FONTE: Compilação dos autores).
De acordo com Chou, Jun & Mansfild (2010), a doença de armazenamento de glicogênio tipo Ia é uma deficiência de glicose-6-fosfatase-α (G6Pase-α). Um complexo funcional de G6PT e G6Pase-α mantém a homeostase interprandial da glicose (Hannah et al., 2022).
“A doença de armazenamento de glicogênio tipo Ia (GSDIa) é causada por variantes bialélicas no G6PC1, levando à perda da função da glicose-6-fosfatase. Como existem déficits tanto de glicogenólise quanto de gliconeogênese, a hipoglicemia emergente é a causa mais significativa de mortalidade precoce.” (BALI et al., 2006)
No GSD tipo I, a capacidade prejudicada de produzir glicose a partir da glicogenólise resulta em hipoglicemia, bem como produção elevada de ácido lático e triglicerídeos (ELLINGWOOD & CHENG, 2018).
ASPECTOS CLÍNICOS
Hipoglicemia
Ao analisarmos a ausência da glicose-6-fosfato no processo da glicogenólise e gliconeogênese em decorrência da irregularidade da glicose-6-fosfatase, podemos verificar que não há como a mesma ser transformada em glicose é assim realizar a travessia pela membrana plasmática. Sendo assim, somente uma pequena parcela, retirada do glicogênio, pode ser utilizada para o controle da glicemia (Berg JM et al, 2004).
Os doentes têm fígado aumentado, retardo do crescimento, por vezes osteoporose, nefromegalia e epistaxis frequentes devido à disfunção plaquetária.
Hiperuricemia
Tendo em vista uma alta taxa de degradação do ATP em resposta à hipoglicemia, nota-se uma baixa disponibilidade de glicose livre, e em decorrência o acúmulo de ADP é transformado em ácido úrico (Yiu WH at al, 2008).
Em decorrência, a hiperuricemia é gerada, assim como Frederico cita:
“A hiperuricemia resulta tanto da diminuição da depuração renal de urato secundária à competição com o ácido láctico e outros, quanto do aumento de produção do ácido úrico” (FREDERICO et al, 2009).
Compilações Renais
Clientes que tem consigo a Glicogenose tipo Ia podem ainda desenvolver complicações da doença, como a hiperglicemia, hiperuricemia, doença renal crônica de origem desconhecida em que a disfunção renal aparece na segunda década de vida do cliente (Yiu WH at al, 2008).
Observa-se também que os clientes com essa anomalia na glicose-6-fosfatase, tem a alteração da aditivação renal e progressão da doença (Yiu WH at al, 2008).
TRATAMENTO
O tratamento da doença de Von Gierke é muito abrangente, realizado principalmente através de dietoterapia para promover a homeostase da glicose e prevenir as reações hipoglicêmicas (Bodinski LH, Ritt R. 2006).
A alimentação através de sondas é uma abordagem tradicional para o tratamento da doença, onde a utilização de polímeros de glicose pela via oral realizam a manutenção dos níveis de glicemia (Reis CVS at al, 2008; Departamento de Nutrição da sociedade brasileira de pediatria, 2004).
O uso do amido de milho cru tem muita eficiência na manutenção da glicemia no corpo, e é recomendado a cada 2 a 4 horas, pois das enzimas amilase pancreática e a glicoamilase intestinal são estimulados e geram a digestão adequada (Reis CVS et al., 2008; Departamento de Nutrição da sociedade brasileira de pediatria, 2004).
O tratamento é muito necessário para que não gere no cliente doenças metabólicas, como graves crises de hipoglicemia que podem levar o cliente a óbito. O tratamento disponível atualmente melhora significativamente a melhora do prognóstico de pacientes que portam essa doença, mesmo sendo relativamente rara (Reis CVS et al., 2008).
CONCLUSÃO
A Glicogenose do Tipo Ia é uma patologia de cunho metabólico hereditário que afeta uma enzima chamada glicose 6-fosfatase que desassocia o fosfato da glicose para manter as concentrações desse monossacarídeo no sangue. Os pacientes com essa patologia acumulam glicose em excesso no fígado, rins e mucosa intestinal, pela inabilidade dessa conversão, desencadeando uma série de achados clínicos e patologias se não tratado. Conhecer os aspectos bioquímicos da doença é de fundamental importância para que se desenvolva um manejo adequado no tratamento e assim, minimizar os seus efeitos deletérios.
A dietoterapia é a pedra angular do tratamento com GSD Ia. A ingestão regular de carboidratos é necessária para prevenir a hipoglicemia e alcançar um bom controle metabólico. O amido de milho é digerido lentamente, assim, pode liberar glicose constantemente entre as refeições. O uso de amido de milho cru em pacientes adultos com GSD1a foi comprovado como um tratamento simples e eficaz a longo prazo.
REFERÊNCIAS
Bali DS, El-Gharbawy A, Austin S, Pendyal S, Kishnani PS. Doença de Armazenamento de Glicogênio Tipo I. 2006 Apr 19 [atualizado 2021 Oct 14]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editores. GeneReviews [Internet]. Seattle (WA): Universidade de Washington, Seattle; 1993–2023. PMID: 20301489.®
Ellingwood SS, Cheng A. Aspectos bioquímicos e clínicos de doenças de armazenamento de glicogênio. J Endocrinol. 2018 Setembro;238(3):R131-R141. DOI: 10.1530/JOE-18-0120. EPub 2018 6 de junho. PMID: 29875163; PMCID: PMC6050127.
Froissart R, Piraud M, Boudjemline AM, Vianey-Saban C, Petit F, Hubert-Buron A, Eberschweiler PT, Gajdos V, Labrune P. Deficiência de glicose-6-fosfatase. Orphanet J Rare Dis. 2011 Maio 20;6:27. DOI: 10.1186/1750-1172-6-27. PMID: 21599942; PMCID: PMC3118311.
Leong, S. Caracterização Molecular, Bioquímica e Clínica de Treze Pacientes com Doença de Armazenamento de Glicogênio 1a na Malásia. Hindawi. https://www.hindawi.com/journals/gr/2022/5870092/.
Olgac A, Okur I, Biberoğlu G, Ezgü FS, Tümer L. Um caso de doença de armazenamento de glicogênio tipo 1a imitando a síndrome de quilomicronemia familiar. Balcãs J Med Genet. 2021 Julho 27;24(1):103-106. DOI: 10.2478/bjmg-2021-0013. PMID: 34447667; PMCID: PMC8366469.
William B. Hannah, Ricardo C. Ong, Margarita Nieto Moreno, Surekha Pendyal, Monica Abdelmalak, Judith Kelsen, Nancy M. McGreal, Priya S. Kishnani, Very early-onset inflammatory bowel disease: Novel description in glycogen storage disease type Ia, Molecular Genetics and Metabolism Reports, Volume 31, 2022, 100848, ISSN 2214-4269, https://doi.org/10.1016/j.ymgmr.2022.100848. (https://www.sciencedirect.com/science/article/pii/S2214426922000088).
1Graduando no Curso de Medicina. Centro Universitário FAMETRO. Manaus-AM. Brasil;
2Mestre e Doutoranda em Biotecnologia. Universidade Federal do Amazonas. Manaus-AM. Brasil.