REGISTRO DOI: 10.69849/revistaft/ra10202501031504
Ana Beatriz de Oliveira Sarmento
Camila Sampaio de Almeida Campinho
Emanuelle da Silva Ferreira
Grayson Amorim Tenório
Jammille Barreto Andrade
Maria Alice Lopes de Almeida
Milena Lyrio dos Santos Joventino
Suallen Amaral Rocha Machado
Valdelino de Jesus Santos Filho
Vanessa Gleyf Sousa Lopes
Docente: Prof.º Marcus Vinicius Cardoso Matos Silva
RESUMO
A Síndrome de Hunter, ou Mucopolissacaridose tipo II (MPS II), é uma doença genética causada pelo acúmulo de glicosaminoglicanos (GAGs) devido à deficiência da enzima iduronato-2-sulfatase (IDS), essencial para a degradação desses compostos. Essa deficiência leva a sintomas progressivos, como deformidades esqueléticas, fácies típicas, problemas cardíacos e hepáticos. Este estudo realizou uma análise in silico de mutações de transversão missense no gene IDS. Foram selecionadas quatro mutações: c.1403G>C, c.230C>A, c.257C>G e c.551G>T, todas associadas a diferentes manifestações clínicas da MPS II. A mutação c.1403G>C causa degradação da proteína IDS, agravando sintomas neurológicos. A c.230C>A está ligada à forma atenuada da doença, afetando principalmente o sistema cardiovascular. A c.257C>G interfere diretamente na função da sulfatase, resultando em uma forma grave da síndrome. A c.551G>T é associada a manifestações precoces e danos aos órgãos vitais. Em todas as mutações analisadas, observa-se comprometimento significativo do sistema nervoso central (SNC), devido ao papel dos GAGs na plasticidade neural e regeneração axonal. A análise estrutural e funcional das mutações revelou que elas alteram a conformação da proteína IDS, prejudicando a degradação dos GAGs e contribuindo para o acúmulo dessas moléculas. A caracterização molecular dessas mutações é fundamental para o desenvolvimento de terapias mais eficazes, como a terapia gênica, visando melhorar o prognóstico e a qualidade de vida dos pacientes com MPS II.
Palavras-chave: Síndrome de Hunter, Mucopolissacaridose tipo II, Gene IDS, Mutações.
ABSTRACT
ABSTRACT: Hunter syndrome, or Mucopolysaccharidosis type II (MPS II), is a genetic disease caused by the accumulation of glycosaminoglycans (GAGs) due to deficiency of the enzyme iduronate-2-sulfatase (IDS), which is essential for the degradation of these compounds. This deficiency leads to progressive symptoms, such as skeletal deformities, characteristic facial features, and cardiac and hepatic problems. This study conducted an in silico analysis of missense transversion mutations in the IDS gene. Four mutations were selected: c.1403G>C, c.230C>A, c.257C>G and c.551G>T, all associated with different clinical manifestations of MPS II. The c.1403G>C mutation leads to degradation of the IDS protein, exacerbating neurological symptoms. c.230C>A is linked to the attenuated form of the disease, mainly affecting the cardiovascular system. c.257C>G directly interferes with sulfatase function, resulting in a severe form of the syndrome. c.551G>T is associated with early manifestations and damage to vital organs. Notably, significant impairment of the central nervous system (CNS) was observed in all analyzed mutations, due to the role of GAGs in neural plasticity and axonal regeneration. Structural and functional analyses of the mutations revealed that they alter the conformation of the IDS protein, impairing the degradation of GAGs and contributing to the accumulation of these molecules. The molecular characterization of these mutations is essential for developing more effective therapies, such as gene therapy, aimed at improving the prognosis and quality of life for patients with MPS II.
Keywords: Hunter syndrome, Mucopolysaccharidosis type II, IDS gene, Mutations.
INTRODUÇÃO
A Síndrome de Hunter, também descrita como Mucopolissacaridose do tipo II é definida pelo acúmulo de glicosaminoglicanos (GAGs), que ocorre em detrimento a deficiência da enzima iduronato-2-sulfatase (IDS), proteína responsável por realizar a catalisação de GAGs. Por conta da deficiência dessa substância, estabelecida por uma falha genética, os polissacarídeos se acumulam em células e tecidos, além de aumentar sua excreção pela via urinária (MOHAMED, 2019). Essas modificações desencadeiam alterações celulares e teciduais irreversíveis que podem culminar em apoptose molecular (D’AVANZO et al., 2020). A expressão fenotípica é abrangente e compreende alterações físicas marcantes como: fácies típica, contraturas articulares, deformidades esqueléticas, pele espessa e alterações que desencadeiam impacto sistêmico como doença cardíaca valvular (MATSUHISA; IMAIZUMI, 2021). Dessa maneira, as manifestações clínicas progridem durante a infância e de tal forma, contribuem para uma redução da qualidade de vida e desenvolvimento cognitivo do paciente (JAZELA-STANEK et al., 2020).
Os achados clínicos supramencionados são encontrados durante uma anamnese bem detalhada e um exame físico que abranja todos os segmentos da propedêutica médica (VERMA et al., 2021). No entanto, a suspeita de diagnóstico de MPS II é confirmada através da análise bioquímica ou genética, além disso a análise qualitativa e quantitativa dos GAGs urinários podem ser usados para triagem (SCARPA et al., 2020). A investigação deve ocorrer, sobretudo, com pacientes do sexo masculino que apresentem um dos sintomas desencadeado pela síndrome (BRASIL, 2024).
A mucopolissacaridose trata-se de uma doença crônica, por isso, os tratamentos disponíveis têm caráter paliativo. Logo, é imprescindível o acompanhamento longitudinal com uma equipe multidisciplinar de saúde, a fim de minimizar os sintomas gerados pela Síndrome de Hunter (ZAPOLNIK; PYRKOSZ, 2021). Existem terapias que diminuem os sintomas da síndrome, como o transplante de células-tronco hematopoiéticas (TCTH) e a terapia de reposição enzimática (TRE) (RACOMA et al., 2021). O padrão atual de cuidado é o uso da terapia de reposição enzimática administrada por infusão intravenosa semanalmente (ZUBER et al., 2023), mas protocolos de terapia gênica também podem ser investigados e aplicados para tratamento (D’AVANZO, 2020). Apesar de já existirem terapias mais recentes, essas foram as que mais demonstraram-se eficazes em reduzir as manifestações clínicas ocasionadas pela doença. Sendo assim, a combinação desses tratamentos pode ser utilizada com intuito de mitigar os sintomas suscitados, e gerar uma melhor qualidade de vida para o indivíduo (D’AVANZO et al., 2020).
O gene alterado nessa síndrome é o iduronato 2-sulfatase, mais conhecido como IDS (RAMÍREZ-HERNÁNDEZ, 2022), também chamado de ID2S, MPS2 ou SIDS (JAZELA-STANEK et al., 2020). O gene IDS consiste de 9 exons, seu locus gênico contém 24 quilobases, está situado no braço longo do cromossomo X (Xq28) (VERMA et al., 2021) e é responsável por produzir a proteína iduronato 2-sulfatase (I2S), uma enzima crucial no processo de degradação do sulfato de heparano e dermatano (D’AVANZO, 2020). Tais enzimas têm papel de sinalização celular e regulação gênica, sendo, portanto, essenciais para o adequado desenvolvimento e funcionamento corporal, mas seu excesso promove muitos prejuízos.
Ademais, a via metabólica associada ao gene IDS é de degradação dos glicosaminoglicanos, mais especificamente as enzimas supracitadas – sulfato de heparano e dermatan – no interior dos lisossomos (MATSUHISA; MAIZUMI, 2021). A expressão do IDS ocorre em diversos tecidos, mas é mais intenso no cérebro, adrenais e pulmões. Assim, as mutações que ocorrem nesse gene levam a acúmulo de glicosaminoglicanos (GAG), defeitos na degradação de GAG, resultando em doenças multissistêmicas complexas (MOHAMED et al., 2019) como a mucopolissacaridose tipo II. Diante do exposto, o presente artigo tem como objetivo analisar as principais mutações gênicas do gene IDS que culminam no desenvolvimento da síndrome de Hunter.
METODOLOGIA
Trata-se de um estudo ecológico in sílico cujo objeto é versado através de uma abordagem explicativa e descritiva. Para tanto, fez-se necessário acessar a National Library of Medicine (NCBI), a fim de obter informações sobre o gene responsável pela Síndrome de Hunter, na plataforma GENE. Em seguida, acessou-se a plataforma ClinVar para buscar as mutações associadas à Mucopolissacaridose do tipo II. Ademais, foram utilizados os sites SciELO e PubMed para subsidiar a criação da introdução e revisão bibliográfica do estudo.
No ClinVar, foram utilizados o descritor booleano IDS AND “MPS II”, resultando na identificação das mutações relacionadas à doença, dos quais foram aplicados os filtros segundo a linha germinativa de ser patogênica, conforme a consequência molecular de ser missense, sendo esta escolhida pela sua maior prevalência e quanto ao o status da revisão foram selecionados os estudos com múltiplas submissões. Segundo a classificação, “status de revisão”, excluiu-se as mutações provavelmente patogênicas, restando apenas às certamente patogênicas que subsidiaram as análises realizadas. Desse total, foram escolhidos os que continham as mutações do tipo transversão, através da identificação do tipo de troca das bases nitrogenadas, apresentada nos estudos selecionados. Somado aos parâmetros descritos, usou-se o critério tempo para selecionar apenas as mutações com pelo menos um artigo publicado após 2020, dessa forma tornou-se necessário a exclusão da mutação c.613G>C, por não atender a este critério da análise, na qual a publicação ocorreu em 1996, restando as mutações: c.1403G>C (artigos de 2012 e 2022); c.257C>G (artigos de 1996 a 2023); c.230C>A (artigos de 2012 a 2022); c.551G>T (artigos de 1996 a 2021). Dentre os artigos mencionados foram excluídos os que apenas citavam as mutações sem descrever implicações relacionadas a sinais e sintomas.
Para a análise quantitativa das mutações identificadas, foram calculadas as frequências absolutas e relativas das diferentes categorias de mutações presentes na amostra estudada, utilizando para isso o programa Microsoft Excel para tabulação e criação de gráficos, facilitando a visualização e interpretação dos dados. Esta análise permite compreender a proporção dos tipos de mutação em relação ao conjunto total, facilitando a identificação das mutações mais prevalentes associadas à Síndrome de Hunter. Foram tabuladas as mutações missense do tipo transversão, certamente patogênicas, através de uma análise clara da distribuição das mutações estudadas.
Os critérios de exclusão adotados neste estudo foram definidos para garantir a qualidade e relevância dos dados. Excluiu-se as mutações que não estavam catalogadas nas bases de dados reconhecidas, como ClinVar ou NCBI, bem como aquelas que não possuíam artigos publicados relacionados ao tema. Outrossim, mutações associadas aos artigos publicados antes de 1996 ou que contavam com apenas uma publicação, também foram descartadas. Dessa forma, buscou-se assegurar a inclusão das mutações com suporte científico consistente e atualizado, conforme descrição no fluxograma abaixo.
Figura 1: Fluxograma para identificação das mutações associadas à Síndrome de Hunter
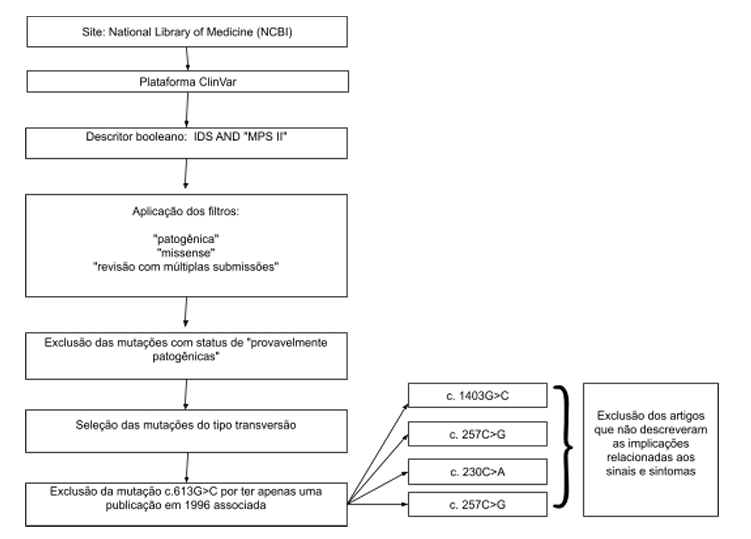
Fonte: elaborado pelo autor (2024)
RESULTADOS E DISCUSSÕES
O gene iduronato 2-sulfatase (IDS) possui 1573 mutações catalogadas na plataforma Clinvar no NCBI, destas 1284 estão associadas à Síndrome de Hunter. A frequência absoluta refere-se ao número total de ocorrência de cada tipo de mutação, sendo 560 patogênicas, das quais 157 são missenses. A frequência relativa foi inferida dividindo-se o número de mutações de cada categoria pelo total de mutações, sendo então expressa em porcentagem, as mutações missenses representam 28% das mutações patogênicas que culminam na Síndrome de Hunter, das quais 3% são transversões e foram analisadas no presente estudo.
Nesse sentido, as mutações missenses são as mais comuns relacionadas a essa síndrome seguidas por nonsense, deleções grandes ou pequenas e rearranjos (RAMÍREZ-HERNÁNDEZ et al., 2022; FULLER; KETTERIDGE, 2021). Dentre estas foram selecionadas apenas as mutações com efeito patogênico que possuem múltiplas publicações atreladas, totalizando 60 mutações. Conforme a classificação “status de revisão”, exclui-se às provavelmente patogênicas, totalizando 12 mutações. Para análise final, foram escolhidas as mutações em que houve transversão, sendo um total de 5 mutações, para comparação da mudança de aminoácido provocada e seus efeitos a nível de sinais e sintomas. Ao ser excluída a mutação c.613G>C por ter apenas uma publicação em 1996, restaram 4 mutações relacionadas, conforme fluxograma citado anteriormente.
Quadro 1: Mutações Missenses de transversão patogênicas atreladas a MPS II
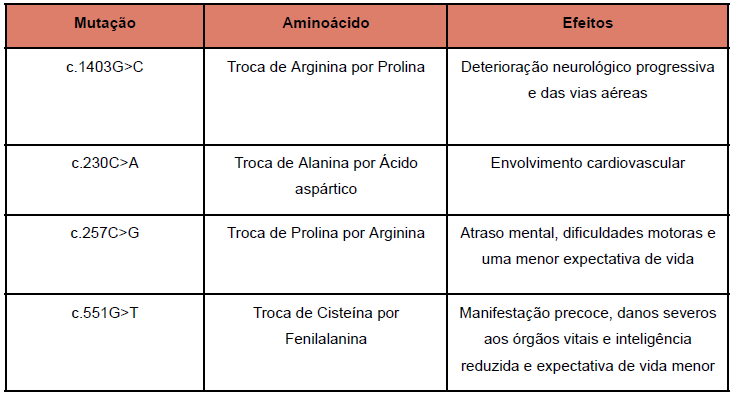
Fonte: Elaborado pelo autor (2024).
Diante do exposto, a mutação missense do gene IDS c.1403G>C (p.Arg468Pro) foi relatado na literatura em indivíduos afetados com Mucopolissacaridose Tipo II (WATTANASATIEN; HERNADEZ, 2022). Essa mutação de transversão, não-sinônima ou missense, que altera a base nitrogenada guanina por citosina localizada no exon 9 no nucleotídeo 1403 (DBSNP, 2024), promove a mudança da categoria química, e resulta em uma alteração não conservadora de aminoácidos na sequência da proteína codificada (BORGES-OSÓRIO; ROBINSON, 2013). Com essa mutação G>C, a arginina, um aminoácido polar é carregado positivamente, é substituído pela prolina, um aminoácido apolar, o que resulta na mudança da interação intramolecular e, dessa forma, na conformação da proteína. Esse defeito no dobramento da proteína ativa o controle de qualidade da célula e quando esse erro não pode ser corrigido, a enzima é retida no retículo endoplasmático e degradada pelo sistema ubiquitina-proteassoma (UPS) (RAMÍREZ-HERNÁNDEZ et al., 2022).
Assim, a alteração estrutural causada pela mutação c.1403G>C implica no ganho de um loop, em vez de uma fita na subunidade SD1, o que causa perda de interação com o ácido pirrolidona carboxílico na posição Q465 (RAMÍREZ-HERNÁNDEZ et al., 2022). Essa modificação conformacional crítica leva ao processo de degradação mencionado e isso reduz drasticamente a quantidade de enzimas funcionais disponíveis nas células, o que impede a degradação adequada dos glicosaminoglicanos (GAGs). Desse modo, promove o acúmulo destes GAGs nos lisossomos e resulta em um fenótipo grave da doença, como observado em pacientes que apresentam essa variante. Tal variante tem uma frequência mundial de <0,01, possui efeito deletério (HERNÁNDEZ et al., 2022) ao passo que torna o gene IDS inativo em células COS7 transfectadas e provoca deterioração neurológica progressiva e doença progressiva das vias aéreas nos pacientes acometidos na forma severa (CHAROENWATTANASATIEN, 2012).
Sucedendo a pesquisa, uma nova mutação missense do gene IDS c.230C>A (p.Ala77Asp) foi relatada em indivíduos com diagnóstico de Mucopolissacaridose Tipo II de forma atenuada (GRINBERG et al., 2012; TORRES et al., 2014). Essa mutação de transversão não-sinônima, que modifica a base nitrogenada citosina por adenina, localizada no nucleotídeo 230, promove a substituição da alanina, um aminoácido neutro e não polar, pelo ácido aspártico, um aminoácido ácido e polar, no códon 77 da proteína IDS que ocasiona uma modelagem avançada da sequência de proteínas e das suas propriedades biofísicas (DBSNP, 2024).
Dessa forma, propõe-se que a mutação sem sentido p.A77D presumivelmente resultou em uma forma branda da Mucopolissacaridose Tipo II ao poupar problemas neurológicos na maioria dos pacientes e quaisquer outros tipos de deficiências graves, devido à atividade enzimática residual ser suficiente por uma expressão reduzida, e não total, da enzima IDS (QUAIO et al., 2012; STEPHAN et al., 2022).
Por conseguinte, a alteração estrutural gerada pela mutação c.230C>A resulta em uma expectativa de vida normal, sendo as complicações mais graves secundárias à defeitos cardíacos, bem como o desempenho cognitivo dentro do padrão. Ademais, o envolvimento cardiovascular relacionado foi citado como a complicação grave mais comum da MPS II devido à insuficiência cardíaca com possibilidade de rápida progressão por um acúmulo dos GAGs no interstício dos miócitos, sendo demonstrado por análise histopatológica do miocárdio. Também foi observado o dano cardíaco devido às valvulopatias, encontradas em até 60% dos pacientes, sendo a valva tricúspide a mais envolvida, com a valva aórtica e mitral afetadas em sequência, respectivamente (STEPHAN et al., 2022).
Além disso, foram reveladas características clássicas da doença como face levemente áspera, hepatomegalia e rigidez articular, especialmente nos ombros, bem como a síndrome do túnel do carpo o que demonstra que as contraturas articulares na MPS II envolvem de forma mais proeminente as extremidades superiores. Outrossim, a obstrução progressiva das vias aéreas foi citada como frequente na doença, sendo necessária correção cirúrgica para hipertrofia dos tecidos adenoides e/ou tonsilares. Houve também casos de retinopatia, embora com menor frequência (QUAIO et al., 2012; STEPHAN et al., 2022). Similarmente, a deficiência quantitativa das células NK e células B também pode estar atrelada a essa mutação, mantendo, no entanto, os valores normais de anticorpos séricos das imunoglobulinas e a resposta padrão aos antígenos polissacarídicos (TORRES LC et al., 2014).
Já a mutação c.257C>G (p.Pro86Arg) no gene IDS é classificada como uma mutação de transversão pois ocorre a mudança da base citosina pela base guanina no DNA (SCHAEFER; THOMPSON, 2014). Tal substituição de bases nitrogenadas ocorre no nucleotídeo 257 localizado no éxons 3 (ZHANG et al., 2019). Com o objetivo de uma ampliação no entendimento dessa mutação, foram desenvolvidos algoritmos para analisar a repercussão de alterações na sequência no splicing de RNA que sugerem que essa variante pode comprometer o sítio de splicing de consenso (DBSNP, 2024).
Assim sendo, tal gene também se classifica como uma mutação não-sinônima ou missense (SCHAEFER; THOMPSON, 2014). Ocorre a substituição do aminoácido prolina, neutro e apolar, pela arginina, que é básico e polar, no códon 86 da proteína IDS (p.Pro86Arg). Resulta-se, desse modo, em uma alteração que tende a ser disruptiva, localizada na Sulfatase, domínio N-terminal (IPR000917) da sequência de proteína codificada, danificando, desta forma, a função da sulfatase de forma bastante direta e resultando em problemas metabólicos graves devido à incapacidade da célula de degradar GAGs corretamente (DBSNP, 2024).
Concomitantemente, a mutação P86R é conhecida por atingir o resíduo de prolina da sequência CXPSR. Sabe-se que a região CXPSR é indispensável para o dobramento proteico e a formação do sítio ativo. A substituição da prolina por outro aminoácido, como arginina, afeta o dobramento correto da proteína. A mutação P86R altera esse resíduo, o que interfere na formação correta do sítio ativo e, por conseguinte, na função enzimática da iduronato-2-sulfatas. Outrossim, além de gerar tais prejuízos supracitados, ocorre também prejuízos no desenvolvimento e na intrínseca propriedade do lisossomo uma vez que nenhuma forma madura lisossomal e nenhum aumento da atividade IDS conseguiram ser detectados (MILLAT et al., 1998). Essa relação evidencia que a mutação c.257C>G resulta, por conseguinte, em uma forma grave de deficiência de iduronato-2-sulfatase, a saber, uma forma grave da síndrome de Hunter.
Ademais, compreende-se que a MPS II é geralmente classificada clinicamente em dois subgrupos: a forma severa e a forma atenuada, de acordo com a abrangência neurológica e o tempo de sobrevida. Dessarte, a forma severa, como a mutação p.Pro86Arg, é a mais frequente e é identificada por um início precoce dos sintomas com comprometimento neurológico gradual como retardo cognitivo, dificuldades motoras e uma significativa diminuição da expectativa de vida. (ZHANG et al., 2019).
Por conseguinte, a mutação c.551G>T (p.Cys184Phe) que ocorre no éxon 5 do nucleotídeo 551 do gene IDS, registrada como CM960862 no banco de dados HGMD resulta em uma mutação missense por transversão que substitui a cisteína por fenilalanina (STENSON, 2024). Essa mudança compromete a função da enzima iduronato-2-sulfatase, responsável pela degradação de glicosaminoglicanos (GAGs), levando ao acúmulo desses compostos e ao desenvolvimento da Mucopolissacaridose tipo II (MPS II). Pacientes com essa mutação frequentemente apresentam a forma grave da doença, com manifestação precoce, danos severos aos órgãos vitais e inteligência reduzida, além de uma expectativa de vida menor (SEMYACHKINA et al.,2021).
Embora seja uma alteração importante, há uma escassez de publicações sobre a mutação c.551G>T, o que limita o entendimento detalhado dos mecanismos moleculares e clínicos envolvidos. Mais estudos são necessários para elucidar seu impacto e melhorar as estratégias de tratamento.
Percebe-se, após a análise das quatro mutações que o acúmulo de GAGs, devido à deficiência da enzima codificada pelo gene IDS, pode ter efeitos significativos no sistema nervoso central, uma vez que esses compostos são essenciais para a plasticidade neural, proliferação celular e regeneração axonal (FOSCARIN et al., 2017). Os proteoglicanos de sulfato de condroitina (CS) e outros GAGs, como sulfato de heparano (HS) e de dermatan (DS), são importantes reguladores na formação de cicatrizes após lesões e na regeneração axonal. Além disso, o sulfato de heparano (HS) tem participação no desenvolvimento do SNC e apresenta característica de estimulador de crescimento dos axônios (REIS, 2013). O acúmulo destes GAGs é responsável pelas condições patológicas associadas à MPS II
(MASHIMA et al., 2016). No contexto de uma deficiência de iduronato-2-sulfatase, o desequilíbrio desses GAGs pode levar a alterações nos padrões de crescimento e regeneração axonal. Assim, uma mutação no gene IDS, que leva ao acúmulo de GAGs como o sulfato de heparano e sulfato de dermatano, pode interferir no desenvolvimento normal do SNC, na capacidade de regeneração após lesões e na homeostase celular.
CONCLUSÃO
A Síndrome de Hunter, ou Mucopolissacaridose tipo II (MPS II), representa um desafio significativo no campo das doenças genéticas raras. Caracterizada pelo acúmulo de glicosaminoglicanos (GAGs) devido à deficiência da enzima iduronato-2-sulfatase (IDS), essa enfermidade leva a manifestações clínicas variadas e progressivas que comprometem severamente a qualidade de vida dos pacientes. A análise genética dessa doença identificou mais de 1.500 mutações no gene IDS, sendo muitas associadas a sintomas graves. Mutações missense, como c.1403G>C, c.230C>A, c.257C>G e c.551G>T, foram destacadas neste estudo, mostrando uma ampla variedade de impactos, desde efeitos neurológicos até cardiovasculares.
Enquanto algumas dessas mutações levam a formas mais graves da doença, outras resultam em sintomas mais leves. A identificação e compreensão dessas mutações têm sido fundamentais para aprimorar o conhecimento sobre a patogênese da MPS II e para impulsionar o desenvolvimento de terapias mais eficazes. As intervenções terapêuticas atuais, como a terapia de reposição enzimática (TRE) e o transplante de células-tronco hematopoiéticas (TCTH), têm caráter paliativo e visam aliviar sintomas. A terapia gênica surge como uma alternativa promissora, porém, sua efetividade clínica ainda depende de avanços significativos na pesquisa e aplicação prática.
A correlação das mutações com os diferentes fenótipos da doença é essencial para o desenvolvimento de tratamentos personalizados. Embora ainda haja escassez de estudos sobre mutações menos comuns e suas consequências clínicas, há esperança de que novas abordagens terapêuticas, especialmente no campo das terapias gênicas e moleculares, possam oferecer opções mais duradouras e eficazes para os pacientes, melhorando seu prognóstico e qualidade de vida.
REFERÊNCIAS
BRASIL. Ministério da Saúde. Terapêuticas: Protocolo clínico e diretrizes da mucopolissacaridose do tipo II, suspeitas clínicas [Brasília]: Ministério da Saúde, agosto 2024.https://www.gov.br/conitec/pt-br/midias/consultas/relatorios/2024/relatorio-preliminar-pc dt-mucopolissacaridose-tipo-ii_cp52. Acesso em: 24 de agosto de 2024.
BORGES-OSÓRIO, Maria R L.; ROBINSON, Wanyce M. Genética humana. Porto Alegre., 3ª edição, p. 56-58: Grupo A, 2013. E-book. ISBN 9788565852906. Disponível em: https://integrada.minhabiblioteca.com.br/#/books/9788565852906/. Acesso em: 12 set. 2024.
CHAROENWATTANASATIEN, Ratana et al. Decreasing activity and altered protein processing of human iduronate-2-sulfatase mutations demonstrated by expression in COS7 cells. Biochemical genetics, v. 50, p. 990-997, 2012.
DREYFUSS, Juliana L. et al. Heparan sulfate proteoglycans: structure, protein interactions and cell signaling. Anais da Academia Brasileira de Ciências, Rio de Janeiro, v. 81, n. 3, p. 409-429, 2009. Disponível em: https://www.scielo.br/aabc. Acesso em: 25 ago. 2024.
D’AVANZO, Francesca et al. Mucopolysaccharidosis type II: one hundred years of research, diagnosis, and treatment. International journal of molecular sciences, v. 21, n. 4, p. 1258, 2020.
DBSNP. Banco de dados de polimorfismos de nucleotídeo único. National Center for Biotechnology Information, 2024. Disponível em: https://www.ncbi.nlm.nih.gov/snp/. Acesso em: 09 set. 2024.
FOSCARIN, S., Raha-Chowdhury, R., Fawcett, J. W., & Kwok, J. C. F. (2017). Brain aging changes proteoglycan sulfation, making perineuronal nets more inhibitory. Aging (Albany NY), 9(6), 1607–1622. Disponível em: https://doi.org/10.18632/aging.101256
FULLER, S.; KETTERIDGE, D. Sobre mecanismos de mutações no gene IDS. Journal of Medical Genetics, 2021. Disponível em: https://jmg.bmj.com/content/early/recent. Acesso em: 25 set. 2024.
JAZELA-STANEK, Aleksandra et al. Diverse clinical outcome of Hunter syndrome in patients with chromosomal aberration encompassing entire and partial IDS deletions: what is important for early diagnosis and counseling?. Clinical Dysmorphology, v. 30, n.2, p. 76-82, 2021.
GRINBERG, Henrique et al. The first cardiac transplant experience in a patient with mucopolysaccharidosis. Cardiovascular Pathology, v. 21, n. 4, p. 358-360, 2012.
HERNÁNDEZ, M.A.; et al. Estudos sobre mutações patogênicas da sulfatase. European Review for Medical & Pharmacological Sciences, 2022. Disponível em: https://www.europeanreview.org/article/2022. Acesso em: 25 set. 2024.
MARTIN, Rick et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics, v. 121, n. 2, p. e377-e386, 2008.
MAROLLA, Ana Paula Cleto et al. Análise da expressão de glicosaminoglicanos sulfatados no tecido humano neoplásico colorretal e na mucosa não neoplásica por espectrometria de massa por ionização por electrospray. Einstein (São Paulo), v. 13, n.4, p. 510-517, 2015. Disponível em: https://doi.org/10.1590/S1679-45082015AO3477. Acesso em: 25 ago. 2024.
MASHIMA, R., Sakai, E., Tanaka, M., Kosuga, M., & Okuyama, T. (2016). Urinary glycosaminoglycan levels in patients with attenuated and severe mucopolysaccharidosis II determined by liquid chromatography-tandem mass spectrometry. Molecular Genetics and Metabolism Reports, 7, 87-93. Disponível em: https://doi.org/10.1016/j.ymgmr.2016.03.009
MATSUHISA, Koji; IMAIZUMI, Kazunori. Loss of function of mutant IDS due to endoplasmic reticulum-associated degradation: new therapeutic opportunities for mucopolysaccharidosis type II. International Journal of Molecular Sciences, v. 22, n. 22, p. 12227, 2021.
MILLAT, G. et al. Estudo sobre a mutação P86R e seu impacto na sulfatase. Journal of Human Genetics, v. 43, p. 285-295, 1998. Disponível em: https://jhg.bmj.com/content/43/3/285. Acesso em: 15 set. 2024
MOHAMED, Shifaza et al. Mucopolysaccharidosis type II (Hunter syndrome): Clinical and biochemical aspects of the disease and approaches to its diagnosis and treatment. Advances in Carbohydrate Chemistry and Biochemistry, v. 77, p. 71-117, 2020.
RACOMA, Marie Julianne C. et al. A review of the clinical outcomes in idursulfase-treated and untreated Filipino patients with mucopolysaccharidosis type II: data from the local lysosomal storage disease registry. Orphanet Journal of Rare Diseases, v. 16, p. 1-16, 2021.
RAMÍREZ-HERNÁNDEZ, M. A. et al. Mutational spectrum of the iduronate-2-sulfatase gene in Mexican patients with Hunter syndrome. European Review for Medical & Pharmacological Sciences, v. 26, n. 14, 2022.
REIS, Evelyn Mendonça. Caracterização dos glicosaminoglicanos do sistema nervoso central e seu envolvimento no processo de neurorregeneração na ascídia Phallusia nigra. 2013. 48 f. Trabalho de Conclusão de Curso (Bacharelado em Farmácia) – Colegiado de Ensino de Graduação – Macaé, Universidade Federal do Rio de Janeiro, Macaé, 2013.
SCARPA, Maurizio et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet journal of rare diseases, v. 6, p. 1-18, 2011.
SEMYACHKINA, A. N., Voskoboeva, E. Y., Nikolaeva, E. A., & Zakharova, E. Y. (2021). Analysis of long-term observations of the large group of Russian patients with Hunter syndrome (mucopolysaccharidosis type II). BMC Medical Genomics, 14, Article 71. https://doi.org/10.1186/s12920-021-00930-3
SOFRONOVA, Viktoriia et al. A Case of Mucopolysaccharidosis II Caused by a Novel Variant with Skin Linear Hyperpigmented Streaks along Blaschko’s Lines. International Journal of Molecular Sciences, v. 24, n. 6, p. 5647, 2023.
STEPHAN, E.; et al. Impactos clínicos das mutações missense na MPS II. Orphanet Journal of Rare Diseases, 2022. Disponível em: https://ojrd.biomedcentral.com/articles/10.1186/s13023-022. Acesso em: 25 set. 2024.
STENSON, P. D. et al. The Human Gene Mutation Database (HGMD®). Cardiff: Institute of Medical Genetics. Disponível em: https://www.hgmd.cf.ac.uk. Acesso em: 08 out. 2024. Referência da mutação: CM960862.
STEPHAN, Bruno de Oliveira et al. Impact of ERT and follow-up of 17 patients from the same family with a mild form of MPS II. Clinics, v. 77, p. 100082, 2022
TORRES, Leuridan Cavalcante et al. NK and B cell deficiency in a MPS type II family with novel mutation in the IDS gene. Clinical Immunology, v. 154, n. 2, p. 100-104, 2014.
QUAIO, C. R. D. C. et al. Report of a large Brazilian family with a very attenuated form of hunter syndrome (MPS II). JIMD Reports-Case and Research Reports, 2012/1, p.125-128, 2012.
VERMA, Shalja et al. A molecular genetics view on Mucopolysaccharidosis Type II. Mutation Research/Reviews in Mutation Research, v. 788, p. 108392, 2021.
WRAITH, J. Edmond et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. European journal of pediatrics, v. 167, p. 267-277, 2008.
ZAPOLNIK, Paweł; PYRKOSZ, Antoni. Gene therapy for mucopolysaccharidosis type II—a review of the current possibilities. International Journal of Molecular Sciences, v. 22, n. 11, p. 5490, 2021.
ZHANG, et al. Classificação clínica e molecular da MPS II. Molecular Genetics and Metabolism, v. 128, p. 105-113, 2019. Disponível em: https://www.sciencedirect.com/science/article/pii/S1096719219301050. Acesso em: 18 set. 2024.
ŻUBER, Zbigniew et al. Diagnosis and management of mucopolysaccharidosis type II (Hunter syndrome) in Poland. Biomedicines, v. 11, n. 6, p. 1668, 2023.
SCHAEFER, G. Bradley; THOMPSON, James. Genética Médica: uma abordagem integrada. AMGH Editora, 2015.
MILLAT, Gilles et al. COS cell expression studies of P86L, P86R, P480L and P480Q Hunter’s disease-causing mutations. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, v. 1406, n. 2, p. 214-218, 1998.
ZHANG, Wen et al. Genetic analysis of 63 Chinese patients with mucopolysaccharidosis type II: Functional characterization of seven novel IDS variants. Clinica Chimica Acta, v. 491, p. 114-120, 2019.