REGISTRO DOI: 10.5281/zenodo.10062817
Karina Negrao Zingra
Caroline Luiza Dias Pereira Bastos
Brunna Yasmin Borges Lérias
Tamiris Lopes Souza Nascimento
Lucas Meira Emerenciano
Lorena Penha de almeida
Mayana Grazielle Souza Vieira
A linfohistiocitose hemofagocítica (LHF) é uma síndrome imunológica rara e potencialmente fatal na qual ocorre ativação descontrolada de linfócitos e macrófagos citotóxicos, causando lesão nos tecidos mediada por citocinas e resulta em disfunções de múltiplos órgãos (GROM, Alexei; HORNE, AnnaCarin; BENEDETTI, Fabrizio, 2016).
Na maior parte dos casos de LHF, principalmente quando em adultos, um gatilho infeccioso, maligno ou iatrogênico está identificado. Portanto, quando a LHF é a aventada hipótese, em pacientes adultos, é obrigatório que os médicos conduzam uma busca minuciosa de gatilhos, guiada pelas características individuais do paciente, dentre elas infecções por Epstein-Barr vírus ou citomegalovírus. (JORDAN, Michael; et al., 2019)
Quanto a epidemiologia a incidência e prevalência de LHF em adultos não é clara, mas uma revisão de Ramos-Casals, 2014, de 2197 casos indicaram idade média de início de 49 anos, maior predominância em homens do que mulheres (1,7:1), e identificou que quase 50% desses casos estão no Japão. Ademais, o mesmo estudo mostra que a mortalidade na LHF em adultos pode chegar a 41% em 1109 adultos diagnosticados. Pode-se concluir que, a LHF é uma síndrome rara, bem pouco reconhecida. Ocorrendo principalmente em recém-nascidos e crianças, e mais raramente adultos. (RAMOS-CASALS, Manoel, et al., 2014; JORDAN, Michael, et al., 2011)
Na classificação atual de distúrbios histiocíticos, a LHF pode ser classificada em LHF primária ou familiar e LHF secundária, também conhecida como adquirida ou LHF reativa.
LHF primária não é uma doença única, mas sim um grupo de distúrbios imunológicos raros, autossômicos recessivos ligados a vários defeitos genéticos, todos afetando a via citolítica mediada por perforina. (GROM, Alexei; HORNE, AnnaCarin; BENEDETTI, Fabrizio, 2016).
Em relação a LHF secundária, esta pode ocorrer em qualquer idade. Em geral, os pacientes tendem a ter apresentações clínicas menos graves do que na LHF primária, mas a mortalidade nesse grupo ainda é considerada alta, e o surgimento dos primeiros sinais clínicos e sintomas geralmente podem estar ligados a uma infecção, em sua maior parte ao Epstein-Barr vírus, citomegalovírus e malignidade. (GROM, Alexei; HORNE, AnnaCarin; BENEDETTI, Fabrizio, 2016).
Quanto ao quadro clínico apresenta-se por um conjunto de sinais e sintomas inespecíficos, de forma aguda ou subaguda, sendo os mais frequentes a febre alta, perda ponderal, astenia, as citopenias, a hepatoesplenomegalia e a presença de hemofagocitose na medula óssea, a linfadenopatia e alterações neurológicas (p. ex., convulsão, hemorragia de retina, ataxia, alteração da consciência ou coma), podendo evoluir rapidamente para a falência multiorgânica e morte.
O diagnóstico mesmo quando há suspeita de LHF, permanece um desafio porque não há achados patognomônicos, clínicos, laboratoriais ou histopatológicos que definam a doença, e ferramentas de diagnóstico atuais carecem de sensibilidade suficiente para serem confiáveis no cenário clínico, especialmente em doença precoce (GRIFFIN, Georgia; SHENOI Susan; HUGHES, Grant, 2020).
Os critérios de diagnósticos de LHF foram desenvolvidos para reconhecer crianças com LHF. (HENTER, Jange-Inge; et al. 2004). Devido à falta de ferramentas melhores, os médico passaram a utilizar esta ferramenta para diagnosticar LHF em adultos. De acordo com os critérios atualizados de LHF em 2004 por Henter, o diagnóstico de LHF requeria cinco das oito características a seguir:
- Febre ≥38,5°C;
- Esplenomegalia;
- Citopenia do sangue periférico, com pelo menos dois dos seguintes: hemoglobina <9 g/dL (para lactentes <4 semanas, hemoglobina <10 g/dL); plaquetas <100.000/microL; contagem absoluta de neutrófilos <1000/microL;
- Hipertrigliceridemia (triglicerídeos em jejum >265 mg/dL) e/ou hipofibrinogenemia (fibrinogênio <150 mg/dL);
- Hemofagocitose na medula óssea, baço, linfonodo ou fígado;
- Atividade de células NK baixa ou ausente;
- Ferritina >500 ng/mL (os autores preferem considerar uma ferritina >3000 ng/mL como mais indicativa de LHF;
- CD25 solúvel elevado (alfa do receptor de IL-2 solúvel [sIL-2R]) dois desvios padrão acima das normas específicas de laboratório ajustadas à idade.
A fim de facilitar esse diagnóstico, em 2014, especialistas criaram um consenso, no qual foi construído um sistema de pontuação projetado para ajudar os médicos no diagnóstico da síndrome hemofagocítica na prática de rotina disponibilizado gratuitamente on-line para facilitar seu uso. Tendo parte destes critérios em conta, foi desenvolvido o Hscore (Tabela 1), com o objetivo de estimar o risco de um doente ter síndrome hemofagocítica (FARDET, Laurence; et al, 2014). O score varia entre 0 e 337, sendo que o cutoff de 169 classifica corretamente 90% dos doentes.
Tabela 1. HScore para estimar risco de um doente ter diagnóstico de síndrome hemofagocítica
Parâmetro Número de pontos Imunossupressão conhecida 0 (não) ou 18 (sim) Temperatura (ºC) 0 (<38.4), 33 (38.4-39.4) ou 49 (>39.4) Organomegalia 0 (não), 23 (hepatomegalia ou esplenomegalia) ou 38 (hepatomegalia e esplenomegalia) Número de citopenias 0 (1 linhagem), 24 (2 linhagens) ou 34 (3 linhagens) Ferritina(μg/L) 0 (<2000), 35 (2000-6000) ou 50 (>6000) Triglicerídeos (mmol/L) 0 (<1.5), 44 (1.5-4) ou 64 (>4) Fibrinogénio (g/dL) 0 (>2.5) ou 30 (≤2.5) Aspartato aminotransferase (U/L) 0 (<30) ou 19 (≥30) Hemofagocitose no aspirado medular 0 (não) ou 35 (sim)
O objetivo deste estudo foi expor um relato de caso da LHF, investigada em um hospital em uma cidade na Amazônia ocidental e mostrar a importância do reconhecimento da síndrome para início mais precoce do tratamento. Será realizada discussão sobre a dificuldade no diagnóstico por ser uma condição clínica rara e com muitos diagnósticos diferenciais que necessitam ser descartados.
DETALHAMENTO DO CASO:
O seguinte caso relatado foi autorizado pela paciente através da assinatura de termo de consentimento livre e esclarecido, encaminhado posteriormente para avaliação e autorização do comitê de ética e pesquisa. Paciente do sexo feminino, 48 anos, iniciou quadro de cefaleia holocraniana em peso associada a mialgia intensa associada a febre de 40 °C. Tais sintomas persistiram após diversas idas ao pronto socorro. Em certo momento foi medicada com penicilina benzatina por suspeita de faringoamigdalite e após 3 dias apresentou rash cutâneo (exantema) difusamente distribuído pela superfície corporal. Na ocasião foi investigado arboviroses incluindo Dengue (IgM negativo e IgG positivo), Chikungunya e Zika, todos negativos. Realizada tomografia de crânio em setor de emergência com evidência de edema cerebral importante. A análise do Líquor estava incolor, límpido, pH 7 e coágulos ausentes, Linfócitos 1/mm3, glicose 59, bacterioscopia sem crescimentos de gram, cloreto 102, desidrogenase láctica (DHL) 22, proteínas totais 23,79, VDRL não reagente, teste rápido não reagente.
Apresentou piora grave dos sintomas sendo encaminhada para unidade de terapia intensiva, onde foi aventada a hipótese de doença de Still do adulto. Após excluídas causas infecciosas e realizado antiparasitário, optou-se por realizar pulsoterapia com metilprednisolona 1g/dia por 3 dias. Paciente referiu melhora significativa das dores musculares e nível de consciência, optado então por manter corticosteroides, recebendo alta da unidade de terapia intensiva e encaminhada ao setor de clínica médica do Hospital de Base Dr Ary Pinheiro para acompanhamento clínico e investigação de demais possibilidades diagnósticas.
Feito rastreio de doenças autoimunes, com auto anticorpos tendo como resultado complemento normal (C3 e C4), fator antinuclear (FAN) não reagente, e todos os outros marcadores (Anti-DNA, fator reumatoide, anti-SM, anti-Ro, anti-La, P-ANCA, C-ANCA) foram negativos. Em investigação de doenças infecciosas apresentava sorologia de Epstein-Barr vírus IgM reagente e IgG reagente, citomegalovírus IgM não reagente e IgG reagente, Hepatite B e C negativos. Quanto aos demais exames apresentava hiperferritinemia (47.767,0 ng/mL), hipertrigliceridemia (490 mg/dL), anemia (Hb: 7,6 mg/dL), neutrófilos (4.678,4/microL), plaquetopenia (22.000/microL), aumento de transaminases (AST 262 U/L e ALT 222 U/L), Proteinúria 24h 2.215,60 mg/24h. Além de características sugestivas de anemia hemolítica com DHL (887 U/L) e coombs direto positivo (IgG 2+), contudo outras provas de hemólise (haptoglobina, reticulócitos, bilirrubina indireta) normais.
Mediante obscuridade do caso e evidência de linfonodos em mediastino demonstrados em tomografia de tórax, foi realizada biópsia do linfonodo na qual exibiu alteração na arquitetura histológica associada a proliferação celular atípica de células médias, e imunohistoquímica mostrou linfonodo exibindo alteração arquitetural a custa de proliferação histiocitária de morfologia usual.
Foi aventada hipótese de LHF e procedido mielograma que representava medula óssea hipercelular às custas de setores granulocítico e megacariocítico com figuras de hemofagocitose presentes (mesmo em uso de corticoide durante toda internação) e aumento de monócitos, em todas as fases de maturação. Achados compatíveis com síndrome de hiperativação macrofágica.
A biópsia de medula óssea não houve evidência morfológica de infiltração neoplásica (linfoma ou histiocitose). Não haviam figuras de hematofagocitose na amostra analisada.
Figura 1. Medula óssea hipercelular às custas dos setores granulocítico e megacariocítico com figuras de hemofagocitose presentes (mesmo em uso de corticóide) e aumento de monócitos, em todas as fases de maturação.
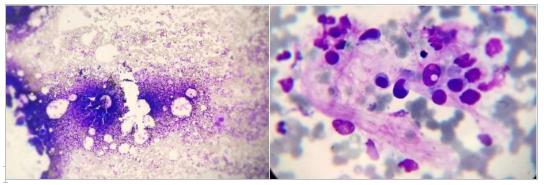
Calculado HScore 241 pontos sugerindo alta probabilidade (>99%) de síndrome hemofagocítica. Após o diagnóstico, foi esclarecido sobre a gravidade da doença e necessidade de tratamento. Como a paciente apresentava programação para mudar de cidade em grande centro, por questões familiares, a mesma optou tratamento no local de nova moradia.
DISCUSSÃO:
Podemos observar no caso que a paciente apresentou inicialmente com quadro febril 40ºC, acompanhado de cefaleia, mialgia e rash cutâneo difuso que poderiam ser justificadas por infecção pelo Epstein-Barr vírus confirmados por exame sorológico. Apesar de a LHF ter gatilhos infecciosos e não infecciosos, a infecção viral é o gatilho mais frequente, seja como infecção primária em pessoas ou após reativação em imunossuprimidos pacientes. Dentre eles, herpes-vírus são responsáveis por até 62% dos casos relatados casos virais de LHF, Epstein-Barr vírus (43%) vírus imunodeficiência humana (22%), citomegalovírus (9%), ,hepatites virais (2,6%, influenza (1,8%), parvovírus B19 (1,8% entre outros.(RAMOS-CASALS, Manoel, et al., 2014).
Outro fato do caso é que a paciente do caso relatado neste trabalho, apresentava níveis importantes de anemia e plaquetopenia. Em mais de 80% dos doentes com LHF é possível identificar citopenias no mínimo em duas de três linhagens hematopoiéticas, isso é explicado pela hemofagocitose direta e pelos aumentados níveis de TNF-α e IFNγ, bloqueiam a hematopoiese. As alterações mais frequente apresenta nos pacientes são anemia (Hb <9mg/dL) e/ou trombocitopenia (plaquetas <100.000/ microL)(ZHANG, Ling, ZHOU, Jun, SOKO, Lubomir, 2014).Também apresentou envolvimento do sistema nervoso central que é uma complicação severa da síndrome hemofagocítica no adulto e afeta entre 20% e 31% dos doentes e principalmente associada a infeção por EBV.
Além disso, apresentou hepatoesplenomegalia alterações cutâneas que podem estar presentes em até 25% dos adultos com LHF, valor ligeiramente inferior ao descrito para a população pediátrica. Sendo que tal característica foi evidenciada no caso relatado neste trabalho. (RAMOS-CASALS, Manoel, et al., 2014).
Reconhecer precocemente a LHF é um dos maiores desafios, especialmente pelo fato que pacientes podem cursar rapidamente com falência de múltiplos órgãos e óbito. Isso porque gera ativação desregulada e proliferação de linfócitos, que desencadeia uma resposta imunológica ineficaz levando à disfunção. Diante de quadro de febre alta e persistente, organomegalia, linfadenopatia, disfunção do sistema nervoso central, disfunção hepática e coagulopatia a hipótese de LHF deve estar dentro do painel de diagnósticos diferenciais. Além dos testes supracitados, podem ser evidenciados laboratoriais indicando valores alterados das transaminases, bilirrubina, lactato desidrogenasse lática (GRIFFIN, Georgia, SHENOI Susan, HUGHES, Grant, 2020; JANKA, Gritta, LEHMBERG Kai)
Logo, a calculadora de probabilidade LHF (HScore) desenvolvida retrospectivamente em pacientes adultos, com parâmetros clínicos e laboratoriais graduados, disponível online, pode ser uma ferramenta de diagnóstico útil e de fácil acesso. (LA ROSÉE, Paul, et al., 2019; ROUPHAE, Nadini, et al. 2007).
Segundo Roldão (2014), o trajeto ideal para chegar ao diagnóstico deve incluir anamnese, exame físico, estudo analítico de exames laboratoriais, pesquisar causas infecciosas como forma de gatilho, estudos com exames de imagem e realização de biópsia ou aspirado de medula óssea. Todos esses passos foram realizados e pormenorizados no estudo do caso da paciente em questão, apesar de nem todas as ferramentas diagnósticas estarem disponíveis na investigação, por falta de recursos. Ainda assim, foi possível realizar o diagnóstico.
Um dado laboratorial que se destaca, é que a paciente mantinha níveis elevados de ferritina sérica, chegando a 47.767,0 ng/mL. A ferritina costuma ser muito elevada especialmente em casos de Doença de Still e LHF e, de fato, o diagnóstico de ambas as síndromes se baseia parcialmente na presença de um nível de ferritina sérica marcadamente elevado. Os critérios diagnósticos para LHF incluem uma ferritina sérica de nível superior a 500 ng/mL. (MOORE, Charles; ORSEMTH, Michelle; FUCHS,Howard, 2013).
A medula óssea é o sítio anatômico preferencial para investigação de suspeita de linfo-histiocitose hemofagocítica, com aspirados positivos identificados em 84% dos adultos relatados casos. A análise da biópsia da medula óssea é menos eficaz (64%) do que os aspirados, mas pode ser útil para descartar neoplasia hematológica subjacente. Ao realizar aspirado de medula óssea na paciente, foi mostrado medula hipercelular com figuras de hemofagocitose, enquanto que a biópsia de medula óssea não mostrou figuras de hemofagocitose, porém também não mostrou infiltração maligna. Vale lembrar que este é um processo fisiológico na qual ocorre fagocitose de células hematopoiéticas por macrófagos ativados e é marcador chave de linfo-histiocitose hemofagocítica, mas não único critério de diagnóstico, devendo avaliado no contexto clínico. (RAMOS-CASALS, Manoel, et al., 2014)
Em um estudo multicêntrico, foram avaliados 312 dos quais 162 pacientes tiveram diagnóstico confirmado de síndrome hemofagocítica. Destes, cerca de 30% dos pacientes com síndrome hemofagocítica não apresentaram características hemofagocíticas em seu aspirado de medula óssea. Por outro lado, mais de um terço dos pacientes sem síndrome hemofagocítica apresentou hemofagocitose na medula óssea aspirada. Mesmo que esses resultados possam parecer confusos, deve-se sublinhar que os quadros de hemofagocitose carecem de especificidade e sensibilidade para o diagnóstico de síndrome hemofagocítica. (RIVIÈRE, Sebástien,et al., 2014)
Após diagnóstico, o protocolo de tratamento da LHF-94, incluindo o etoposido, tem alta eficácia no tratamento do excesso de inflamação em adultos com HLH sendo altamente recomendado. Tal protocolo consiste em corticosteróides, geralmente dexametasona, ciclosporina A (CSA), terapia intratecal e etoposido, para deletar células T ativadas e suprimir a produção de citocinas inflamatórias. Para a paciente foi iniciado pulsoterapia com dexametasona, contudo, foi transferida para seguimento de protocolo em outra cidade devido questões familiares. Apesar do tratamento contínuo da LHF, é extremamente recomendado que o rastreio de outros gatilhos de LHF, como malignidades ocultas sejam investigados profundamente (LA ROSEE, Paul, et al., 2019)
Foi realizada uma revisão sistemática por KNAAK et al. (2020), No total, 661 pacientes de 65 estudos e casos séries foram incluídas. Entre os pacientes com um gatilho infeccioso, 77 (23,3%) não receberam tratamento específico para LHF, dos quais 38 (49,4%) sobreviveram. Isso sugere que eliminar o gatilho é de extrema importância para a sobrevivência e, em alguns casos selecionados, suficientes para tratar LHF. Assim, a necessidade de imunossupressores podem ser reduzidos em pacientes onde a infecção está desencadeando LHF.
Na maioria dos estudos, a taxa de mortalidade foi alta, principalmente no primeiro mês após o diagnóstico. Além disso pacientes com malignidades hematológicas tiveram uma sobrevida de curto prazo pior do que pacientes com síndrome hemofagocítica associada à infecção. Ainda assim, apesar da evolução do reconhecimento e tratamento da síndrome hemofagocítica, a mortalidade no adulto é bastante elevada e está reportada entre os 42% e 75%.(RIVIÈRE, Sebástien,et al., 2014; LI Fei, et al, 2015).
KNAAK (2020), em sua revisão sistemática mostrou que a LHF pode alcançar taxas de mortalidade de até 57,8%, quando fator desencadeante não é identificado até 75,8% e menor em LHF associada a autoimune (36,3%). As infecções, nesse estudo, mais uma vez foram os gatilhos mais frequentes em LHF gravemente doente pacientes (49,9%).
No estudo de Li (2015), para verificar quais fatores clínicos ou laboratoriais específicos em diagnóstico previu o prognóstico de pacientes com LHF, analisaram os dados de sobrevida dos 85 pacientes com LHF. Ao reunir os resultados, estes revelaram que pacientes adultos com LHF tinham um espectro clínico variável, bem como doenças subjacentes. Pacientes com infecção ativa por EBV e linfoma tiveram pouca sobrevivência. Para encontrar alguns indicadores para predição do risco de morte em pacientes com LHF, foi comparada a diferença de vários indicadores laboratoriais entre pacientes sobreviventes e mortos. Não houve diferença entre muitos dos indicadores incluindo glóbulos brancos, hemoglobina, ferritina sérica, transaminases, bilirrubinas e triglicerídeos. No entanto, no mesmo estudo, tiveram alta mortalidade bem como sobrevida inferior se houvesse: fibrinogênio <1,5 g/L, plaquetas <40.000 /microL, e DHL ≥1000 U/L.
CONCLUSÃO:
Este trabalho foi desenvolvido a fim de evidenciar a provação necessária para realizar o diagnóstico da LHF. Foi relatado o caso de uma mulher de meia idade no qual provável gatilho tenha sido infecção pelo EBV. Sabe-se que mais comum em homens, o que evidencia a raridade do caso descrito, e os gatilhos infecciosos por infecções virais na LHF secundária são comumente relatados nos trabalhos descritos sobre tal patologia.
A LHF é uma doença que deve ser sempre lembrada em pacientes com febre de origem obscura, especialmente com plaquetopenia associada a lesão de outros órgãos. O mielograma, bem como a análise de lesão hepática, triglicerídeos e ferritina devem estar inclusos na primeira parte da investigação. Mediante o aumento de histiócitos e células fagocíticas deve ser feita minuciosa procura de infecção ou malignidade subjacente.
A celeridade no diagnóstico permite início prematuro do tratamento, baseado na causa inicial. Isso pode evitar evolução extremamente ruim, com alta agressividade e mortalidade elevada nos pacientes acometidos. É de grande preocupação que a LHF costuma ser subdiagnosticada ou não é possível fechar todos os critérios. Sendo assim, se houver suspeita diagnóstica o tratamento incluindo corticosteroides e quimioterápicos deve ser instituído, além de terapia antiviral, antifúngica e antibacteriana de amplos espectros.
REFERENCIAS
FARDET, Laurence; et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol.Vol. 66 n.9p.2613-2620,2014
GRIFFIN, Georgia, SHENOI Susan,HUGHES, Grant. Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy. Best Practice & Research clinical rheumatology. Vol. 34, N. 4, Agosto, 2020.
GROM, Alexei; HORNE, AnnaCarin; BENEDETTI, Fabrizio. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol vol. 12 N. 5.p 259-268, Maio,2016.
HENTER, Jange-Inge; et al. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatric Blood Cancer, Vol. 48, p. 124–131, 2004.
RAMOS-CASALS, Manoel, et al. Adult haemophagocytic syndrome. Lancet Vol. 383: p. 1503–1516, Abril, 2014.
JANKA, Gritta, LEHMBERG Kai. Hemophagocytic syndrome – an update. Blood Reviews, vol. 28 n.4, p. 135-142, Julho, 2014
JORDAN, Michael B., et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from The North American Consortium for histiocytosis (NACHO). Pediatric Blood Cancer;Vol. 66 n. 1 e.27929, Novembro, 2019
JORDAN, Michael B. et al. How I treat hemophagocytic lymphohistiocytosis. Blood First Edition paper. Vol. 118 n. 15, p. 4041-4052, 2011.
KNAAK, Cornelia, et al. Treatment and Mortality of Hemophagocytic Lymphohistiocytosis in Adult Critically Ill Patients: A Systematic Review With Pooled Analysis. Critical Care Medicine, vol. 48, n. 11, p. e1137-1146, Novembro, 2020.
LA ROSÉE, Paul, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. The American Society of Hematology. Vol. 133, n. 23, p. 2465-2477, 2019.
Song Y, Pei RJ, Wang YN, Zhang J, Wang Z. Central Nervous System Involvement in Hemophagocytic Lymphohistiocytosis in Adults: A Retrospective Analysis of 96 Patients in a Single Center. Chin Med J (Engl). 2018;131(7):776-783
LI Fei, et al. Clinical characteristics and prognostic factors of adult hemophagocytic syndrome patients: a retrospective study of increasing awareness of a disease from a single-center in China. Orphanet Journal of Rare Diseases, Vol. 10, n. 20, p. 1-9, 2015.
MOORE, Charles; ORSEMTH, Michelle; FUCHS,Howard. Causes and significance of markedly elevated serum ferritin levels in an academic medical center. Journal of Clinical Rheumatology, Sep; vol. 19 n. 6, p. 324-328, setembro, 2013
RIVIÈRE, Sebástien,et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. The American Journal of Medicine. Vol. 127 n. 11, p. 1118-1125, Abril, 2014
ROLDAO, Marisa. Linfohistiocitose Hemofagocítica Primária e Secundária: fisiopatologia, diagnóstico e tratamento. 2017. Número de folhas ou volumes. Mestrado integrado em Medicina – Faculdade de medicina de Lisboa, Lisboal,2017.
ROUPHAE, Nadini, et al. Infections associated with haemophagocytic syndrome. Lancet Infectous Disease. vol.7 n.12 p. 814-822, 2007.
VOLPATO, Fabiana; YASSUDA FILHO, Paulino. Hemolytic anemia supposedly related to hematophagocytic syndrome in a child attended at the University Hospital of the West of Paraná (HUOP). Revista Brasileira de Análises clínicas, vol. 51, n. 3, p. 253-256, 2019.
ZHANG, Ling, ZHOU, Jun, SOKO, Lubomir. Hereditary and acquired hemophagocytic lymphohistiocytosis. Cancer Control, vol. 21, n. 4 p. 301-312, 2014
Orientadora: Dra Mayana G. Souza Vieira
Hospital De Base Dr. Ary Pinheiro – Porto Velho – RO