TUBEROUS SCLEROSIS: CASE REPORT
REGISTRO DOI: 10.69849/revistaft/ch10202410312018
Juliana Faleiro Pires1;Giovanna Maria Ribeiro Silva2; Ekaterine Apostolos Dagios3; Eduardo José Ferreira Sales4; Ana Carolina Buta Pereira5; Heloise Fernandes da Silva Bastos6; Ana Couto Lima7; Vitória Maria Fulanette Corrêa8; Renata Machado Bonfim9; Luciany Almeida de Carvalho10
Resumo
INTRODUÇÃO: A esclerose tuberosa (ET) é uma patologia rara, multissistêmica e de caráter autossômico dominante, causada por mutação do tipo deleção nos genes de supressão tumoral TSC1 e TSC2 RELATO DE CASO: Trata-se de criança de 5 anos que nos primeiros dias de vida devido presença de sopro cardíaco foi indicado a realização de Ecocardiograma, o qual evidenciou a presença de múltiplas massas intracardíacas em ventrículos direito e esquerdo. Após exame foi orientado investigação de tumores em outras áreas do corpo, devido à alta relação dos rabdomiomas cardíacos com Esclerose Tuberosa (ET). Na TC de crânio foi visualizado túberes corticais e nódulos harmatosos subependimários e fechado diagnóstico de ET. No momento, a criança em seguimento multidisciplinar, segue bem mas com piora das lesões em Ressonância Magnética (RNM) de Crânio. DISCUSSÃO: A ET pode ser uma doença devastadora, com grande morbidade. Dessa maneira, é importante diagnosticar precocemente. O acompanhamento multiprofissional é capaz de melhorar o prognóstico da doença, melhorando a qualidade vida e sobrevida destes pacientes.
Palavras-chaves: esclerose tuberosa, hamartoma, epilepsia, aconselhamento genético, equipe multiprofissional
INTRODUÇÃO
A esclerose tuberosa (ET) é uma patologia rara, multissistêmica e de caráter autossômico dominante, causada por mutação do tipo deleção nos genes de supressão tumoral TSC1 e TSC2. Ambos são responsáveis pela codificação das proteínas tuberina e hamartina. Os casos familiares estão associados a mutação no TSC1, e as mutações germinativas e de maior gravidade relacionadas ao TSC2. A doença possui diversas apresentações clínicas e pode ser diagnosticada por meio de testes genéticos.O complexo tuberina-hamartina fisiologicamente inibe a rota da rapamicina (mTOR). A via do alvo da rapamicina em mamíferos (mTOR) é uma via de sinalização celular que envolve funções fisiológicas importantes, incluindo crescimento celular, proliferação, metabolismo, síntese de proteínas e autofagia. Assim, com os loci mutados, ocorre uma hiperativação do mTOR, o que resulta em diferenciação anômala das células do organismo e formação de hamartomas em diversos sistemas, como nos olhos, pele, rins, coração e cérebro. (MONICH, A.G., Bissler J.J., Barreto, F,C, 2024)
A ET apresenta manifestações clínicas variáveis, com acometimento cutâneo, pulmonar, renal, cardíaco e cerebral. O desenvolvimento de lesões dermatológicas é a manifestação clínica mais comum. O quadro é marcado por manchas hipocrômicas e acrômicas, afetando de 90 a 98% dos pacientes, principalmente antes da puberdade. As lesões do sistema nervoso podem estar presentes em 80% dos pacientes e são as principais causas de morbimortalidade. (PORTOCARRERO LKL.,et al., 2018)
Este artigo possui o objetivo de relatar o caso de uma criança diagnosticada precocemente com ET, correlacionando os achados e a conduta clínica com os dados da literatura.
RELATO DE CASO
RN, termo, AIG, Capurro 38 semanas e 4 dias, sexo feminino, nascida de parto cesariano devido Hipertensão Arterial Sistêmica materna, realizou 8 consultas de pré-natal, sem alterações sorológicas e de outros exames complementares. Nasceu com Apgar 9/9, líquido amniótico claro e sem sinais de sofrimento fetal. A paciente evoluiu com desconforto respiratório após 2 horas de vida, sendo indicado CPAP. Nas primeiras 24 horas de vida, evoluiu com melhora do padrão respiratório, entretanto, permaneceu hiporreativa e com dificuldade no aleitamento materno. Os rastreios infecciosos e hemoculturas foram negativos e não houve alteração no Teste do Coraçãozinho.
No terceiro dia de vida, ao exame físico foi auscultado sopro cardíaco e então solicitado Ecocardiograma (ECO). O qual demonstrou múltiplas massas intracardíacas em ventrículos direito e esquerdo, tanto intracardíacas como intramurais. Observadas duas em via de saída de ventrículo direito mas sem causar obstrução significativa ao fluxo. Outras em folheto anterior da valva mitral sem disfunção desta associada. Observado durante exame FC limítrofe em torno de 80-90 bpm. Solicitado após Eletrocardiograma que demonstrou: ritmo sinusal com FC 88bpm com extrassístoles ventriculares. Por fim, os achados foram sugestivos de rabdomiomas, mas com evolução clínica atual estável, sem sinais de baixo débito sistêmico ou pulmonar.
Figura 1: Imagem apical quatro câmaras demonstrando a presença de vários tumores em ambos ventrículos, especialmente em ventrículo direito.
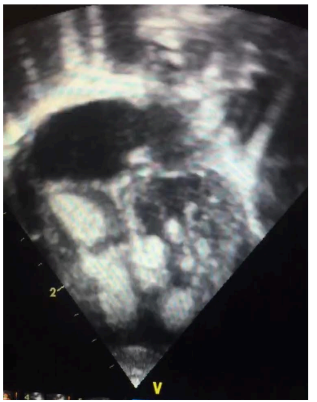
Fonte: arquivo pessoal
Figura 2: Imagem apical da via de saída de ventrículo esquerdo demonstrando a presença de vários tumores especialmente em região septal.
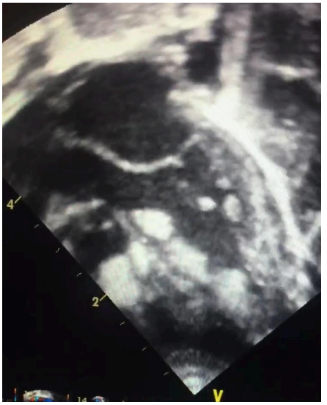
Fonte: arquivo pessoal
Figura 3: Imagem subcostal demonstrando via de saída de ventrículo direito com vários tumores nesta região.
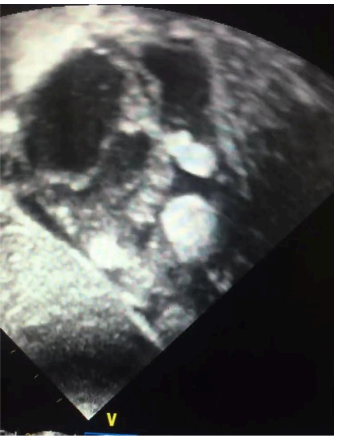
Fonte: arquivo pessoal
Figura 4: Via de saída do ventrículo direito, demonstrando presença de tumores nesta região mas sem comprometimento ao fluxo.
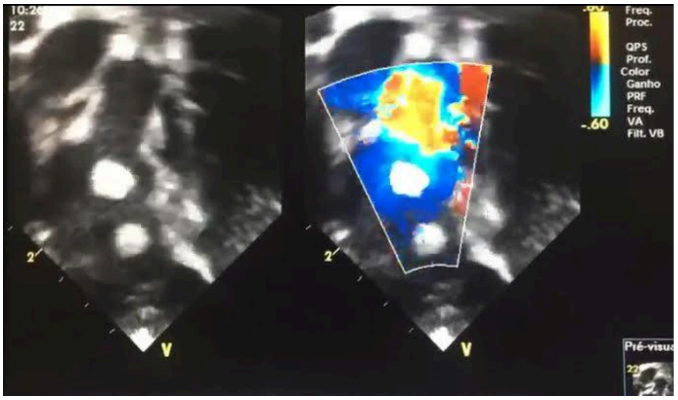
Fonte: arquivo pessoal
Figura 5: Imagem subcostal, aos 5 anos, demonstrando a presença de tumor no ventrículo direito.
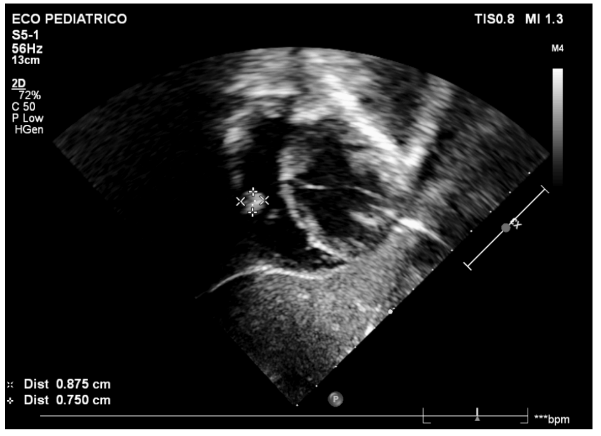
Fonte: arquivo pessoal
Figuras 6 e 7: Imagens em corte apical em quatro câmaras, aos 5 anos, demonstrando a presença de tumores no ventrículo direito mas com redução de outros presentes no período neonatal.
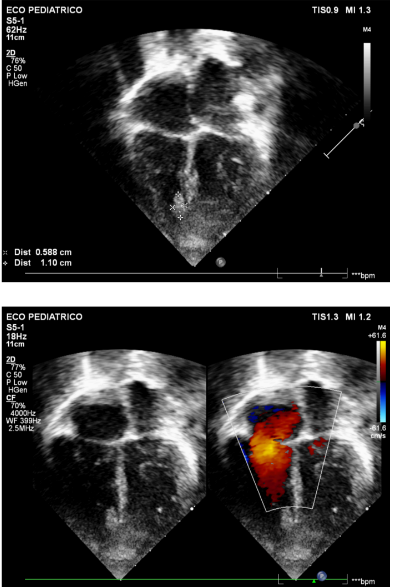
Fonte: arquivo pessoal
Figuras 8 e 9: Imagens em corte apical cinco câmaras, aos 5 anos, demonstrando fluxo preservado em via de saída de ventrículo esquerdo e redução na presença dos tumores observados previamente em região septal.
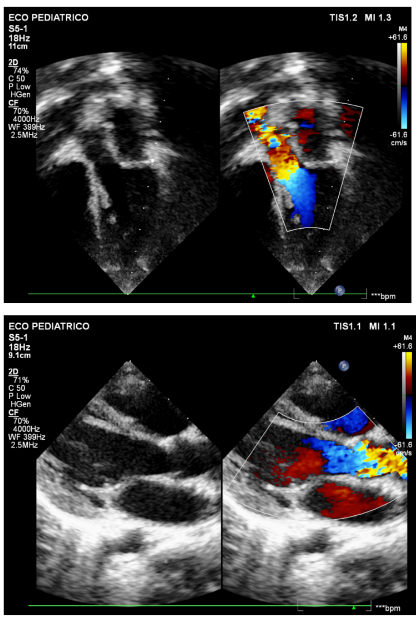
Fonte: arquivo pessoal
Figura 10: Imagem em corte paraesternal eixo curto, aos – anos, demonstrando fluxo preservado em via de saída de ventrículo direito não sendo mais observados tumores presentes nesta região no período neonatal.
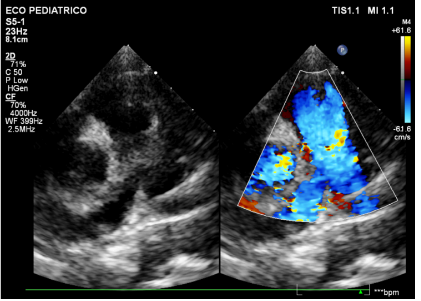
Fonte: arquivo pessoal
Durante a internação hospitalar também foi notado em exame físico inúmeras (>10) manchas hipocrômicas pelo corpo, principalmente em abdome, tórax e membros. Diante da alterações dermatológicas e da presença de rabdomioma em ECO, foi orientado investigação de tumores em outras áreas do corpo, devido à alta relação dos rabdomiomas cardíacos com Esclerose Tuberosa (ET). Para isso, foi solicitado tomografia computadorizada (TC) de crânio, fundoscopia, ultrassom (USG) de abdome e Holter de 24 horas.
Figuras 11,12,13 e 14: máculas hipocrômicas
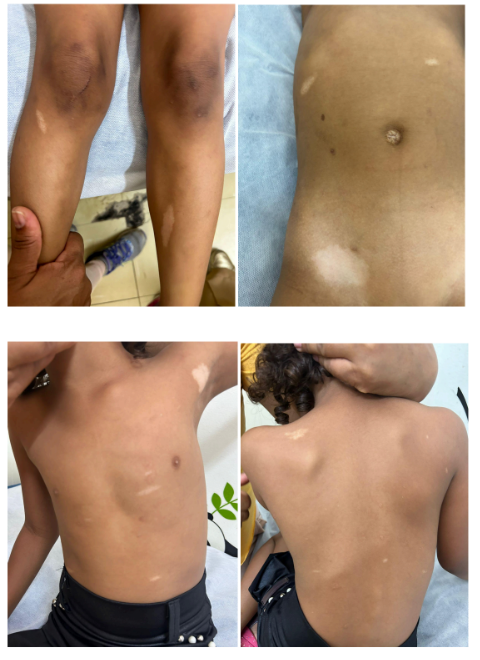
Figura: arquivo pessoal
A fundoscopia e USG de abdome total, sem alterações. Entretanto, foi demonstrado na TC de crânio sem contraste imagens grosseiramente arredondadas hiperdensas intraparenquimatosas (inferior a densidade hemática aguda) nos lobos frontal e parietal esquerda, bem como subependimários adjacentes ao corpo e átrio do ventrículo lateral direito e dos cornos frontais, sendo esses dois os maiores e relativamente simétricos, medindo até 6mm. Dessa forma, no contexto de suspeita clínica de ET, esses achados podem ser considerados como túberes corticais e nódulos harmatosos subependimários da referida doença. No Holter de 24 horas, apresentou durante toda a gravação arritmia sinusal fásica, determinando variação brusca da FC (78-178 bpm). 3639 extrassístoles supraventriculares isoladas. 17 extrassístoles ventriculares isoladas. Sem alterações de segmento ST.
Durante a internação não houve relato de crises epilépticas. Ao exame neurológico estava sem alterações de força e tônus e os reflexos estavam preservados. EEG normal. A paciente recebeu alta hospitalar aos 17 dias de vida, com seguimento ambulatorial.
Aos 7 meses, a mãe referiu que a paciente iniciou episódios convulsivos, associado com redução na força muscular à direita. Episódios convulsivos caracterizados por choro intenso, seguido de hipertonia generalizada e cianose perioral, evoluindo após com hipotonia generalizada e sonolência e então iniciado fenobarbital. Em retorno aos 9 meses de idade, a mãe referiu que a criança não apresentou novos episódios de crises convulsivas. Em EEG de controle realizado aos 11 meses de idade, foi relatado alentecimento intermitente temporal direito.
Aos 9 meses em Holter de controle, foi identificado bradicardias patológicas com esboço de onda delta com intervalo PR curto, sugestivo de Wolff-Parkinson White (WPW). Ainda, houve registro de 8 extrassístoles ventriculares. Em consulta com a cardiologista, foi iniciado amiodarona, uma vez que a criança apresentava episódios de cianose labial e perda da consciência, podendo estar associada a WPW. Em retorno após o início da medicação, a mãe negou novos episódios de cianose ou perda da consciência. Após alguns meses, a amiodarona foi trocada pelo propranolol, por apresentar menos efeitos colaterais. Paciente permaneceu sem sintomas.
Em junho de 2020, a paciente realizou microcirurgia para retirada de tumor intracraniano temporo-parietal. Não houve complicações cirúrgicas e a paciente recebeu alta 4 dias após o procedimento. Entretanto, aos 1 ano e 10 meses, a paciente voltou a apresentar crises convulsivas, mesmo com as medicações em uso. Ainda, houve piora progressiva das crises, sendo a última mais intensa e demorada do que as anteriores.
Figura 15: Corte axial de ressonância magnética, sequência FLAIR, evidenciando lesões corticossubcorticais nos lobos frontais (setas brancas), sugestivas de túberes corticais.
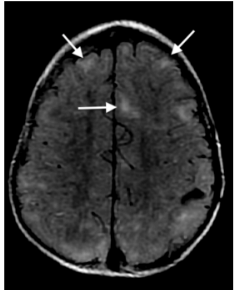
Fonte: arquivo pessoal
Figura 16: Corte axial de ressonância magnética, sequência FLAIR, evidenciando nódulos subependimários (setas brancas), prováveis hamartomas, e cavidade cirúrgica (cabeça de seta).
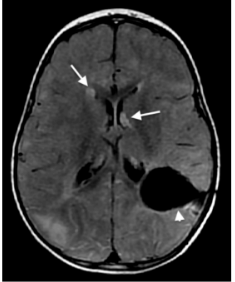
Fonte: arquivo pessoal
Aos 2 anos, a paciente ainda apresentava crises convulsivas atônicas, sem espasmos ou hipertonias. Mãe relatava cianose labial e negava liberação esfincteriana ou sialorréia durantes as crises. Ademais, as crises duravam aproximadamente 10 minutos, no máximo, e aconteciam por volta de 1 vez ao mês. Assim, foi realizada nova microcirurgia em março de 2021 para ressecção de resíduo do tumor cerebral em região temporal esquerda. Após a cirurgia, o fenobarbital foi suspenso e foi iniciada a carbamazepina. Dessa forma, os sintomas foram controlados e a paciente evoluiu sem novas crises epilépticas.
Aos 3 anos, a paciente começou a apresentar sintomas sugestivos do transtorno do espectro autista (TEA) e foi prescrito risperidona e iniciado seguimento com a Psiquiatria e Psicologia. Aos anos iniciou seguimento com a Nefrologia devido alteração em Ultrassom renal e vias urinária feito em março de 2023, que evidenciou numerosas imagens nodulares e ovais, hiperecogênica e circunscritas na cortical medindo a maior 2mm, sem vascularização ao doppler. Imagens sugestivas de angiolipomas renais, apesar de função renal normal.
Figuras 17 e 18: Imagens de cortes longitudinais dos rins direito e esquerdo, evidenciando alguns dos incontáveis nódulos hiperecogênicos (localizados entre os calipers) visibilizados nas corticais renais, que mais provavelmente correspondem a angiomiolipomas.
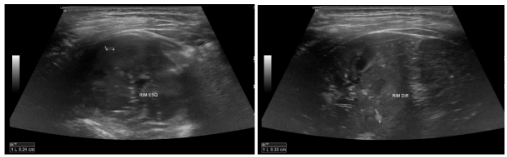
Fonte: arquivo pessoal
Em novembro de 2023, realizou nova RNM de crânio, na qual foi encontrado aumento do nódulo sólido em comparação ao exame do ano anterior, com intenso realce no corno frontal do ventrículo lateral esquerdo, adjacente ao forame de Monro, que promove leve compressão do septo pelúcido e do ventrículo lateral direito. Mede 26 x 20 x 18 mm (medida anterior 15 x 11 x 12 mm). Os achados sugerem astrocitoma subependimário de células gigantes. Persistência dos múltiplos nódulos subependimários com realce pelo meio de contraste, alguns calcificados, o maior no corpo do ventrículo lateral direito, medindo 8 x 5 mm. Lesões córtico-subcorticais com hipersinal T2/FLAIR, algumas com tênue hipersinal T1, esparsas nos hemisférios cerebrais, sugestivos de túberes corticais.A nova abordagem cirúrgica ainda está sendo planejada.
Figuras 19 e 20: Corte axial de ressonância magnética, sequência T1 pós-injeção de gadolínio, e corte coronal, sequência T1 com saturação de gordura pós-injeção de gadolínio, demonstrando aumento do nódulo subependimário próximo ao forame de Monro esquerdo (setas brancas), evidenciado em exames anteriores, medindo 2,5 cm no atual estudo, sugestivo de astrocitoma subependimário de células gigantes.
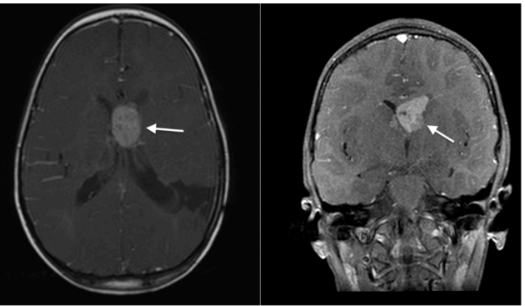
Fonte: arquivo pessoal
Em última consulta com a Oncologia Pediátrica foi optado por iniciar o Everolimus 3 mg/dia devido surgimento de angiolipomas renais e aumento da lesão no Sistema Nervoso Central. Entretanto, a medicação é de alto custo e ainda não foi liberada para o uso da paciente.
DISCUSSÃO
A ET é uma doença considerada rara e sua incidência é estimada em 1 a cada 6 a 10 mil nascidos vivos. Os maiores guidelines internacionais sobre o manejo da ET foram publicados em 1998, 1997, 2013 e, por fim, atualizados em 2021. As manifestações da doença podem estar associadas a diferentes níveis de morbidade, inclusive quadros graves que podem levar à morte. Portanto, o diagnóstico precoce, em conjunto com o acompanhamento ambulatorial, é essencial no manejo da doença. Na maioria dos casos os pacientes acometidos pela ET procuram atendimento devido a convulsões ou queixas dermatológicas. Ambos os sexos são igualmente afetados, porém as meninas possuem quadro clínico mais acentuado. (NORTHRUP, H., et al., 2021)
As lesões dermatológicas são as mais comuns, e acometem cerca de 90-98% dos pacientes. Dentre as manifestações cutâneas, as máculas hipopigmentadas estão presentes em 90% dos casos, são encontradas ao nascimento e permitem diagnóstico precoce. Enquanto que os angiofibromas faciais podem ser observados em cerca de 80-90% dos casos, aparecem no 3 a 4 ° ano de vida da criança, podem piorar na adolescência e estabilizar-se na fase adulta. Os angiofibromas são caracterizados por pápulas, principalmente em região malar, dorso nasal, fronte, queixo, sulco nasolabial, e com cor variável. (PORTOCARRERO LKL.,et al., 2018) Na paciente em questão desde o nascimento foi notado presença de manchas hipocrômicas em região de abdome, tórax e membros, o que corrobora com a literatura, por se tratar da alteração mais frequente nos paciente com ET.
Do ponto de vista pulmonar, há uma substituição do tecido alveolar por cisto e musculatura lisa, denominada linfangioleiomiomatose (LAM), com incidência de 40%, surge quase que exclusivamente nas mulheres. Na LAM, as células musculares sofrem um crescimento anormal, de modo que a elasticidade pulmonar vai reduzindo, diminuindo assim a capacidade vital e aumento do volume residual. Os sintomas mais comuns são: tosse, dispneia e hemoptise. Em relação ao aparelho cardiovascular, o rabdomioma é o tumor cardíaco mais comum, aparece de forma precoce no feto e no recém-nascido. Na maior parte dos casos é assintomático, porém a depender da localização pode evoluir com cardiomegalia, sopro, arritmias, alteração do fluxo sanguíneo e óbito. Sendo que por vezes o diagnóstico é feito durante o pré-natal. (PORTOCARRERO LKL.,et al., 2018). Durante a internação da paciente na Unidade de Cuidados Intermediários Neonatais, foi notada presença de sopro cardíaco em exame físico. Foi então solicitado Ecocardiograma que evidenciou a presença múltiplos tumores intracardíacos sugestivos de rabdomiomas sem sinais de baixo débito sistêmico ou pulmonar. E foi a partir desse exame que foi iniciada a investigação para ET. O que demonstra a importância de exames complementares para o diagnóstico precoce, a fim de que a criança inicie o acompanhamento multiprofissional e assim evitando futuras complicações.
A epilepsia acomete 70 a 90% dos pacientes, geralmente acontecem nos primeiros 3 anos de vida. Os pacientes podem apresentar qualquer tipo de convulsão, sendo as convulsões focais e espasmos epiléticos os mais comuns. Os exames de imagem geralmente apresentam tubérculos subcorticais, linhas de migração na substância branca, nódulos subependimários na parede lateral do ventrículo e em 3° ventrículo em 80% dos pacientes, e também astrocitomas subependimários de células gigantes em 5 a 15% dos casos. Esta última lesão a depender do tamanho pode complicar com ventriculomegalia e hidrocefalia. Além das lesões estruturais, os pacientes podem apresentar Transtorno do Espectro Autista e problemas comportamentais em 40 a 50% dos casos, déficit de atenção e hiperatividade em 30 a 40% dos pacientes. (PEREIRA CCS., et al.,2022) (NORTHRUP, H., et al., 2021). Diante das alterações em Ecocardiograma e alterações de pele, foi solicitado TC de crânio, o que evidenciou a presença de túberes subcorticais e nódulos subependimários, tais alterações são comuns e ocorre em 80% dos casos, o que reforça as dados da literatura. A criança em questão foi submetida a duas microcirurgias para retirada de tumor intracraniano e está em uso de carbamazepina para controle das crises epilépticas. Em última RNM de crânio, foi visualizado presença de astrocitoma subependimário de células gigantes, tais lesões são menos comuns e associadas a complicações. A criança aguarda nova cirurgia e está em seguimento com a Neurologia e Neurocirurgia.
Em relação a parte renal, os angiomiolipomas são tumores benignos comuns no paciente com ET, sendo 80% dos casos. Os quais têm início nos primeiros anos de vida e com crescimento mais evidente durante a adolescência ou início da fase adulta. A maioria é assintomática, porém apresenta morbimortalidade, associado a quadros de hematúria, perda de função renal e risco de hemorragia. O paciente com ET também tem maior incidência de múltiplos cistos renais, em relação à população geral. E por fim, há relatos de associação com carcinoma de células renais, considerado raro, e com incidência global de 1 a 2%. (MONICH, A.G., et al., 2024). No caso em questão, após aventada hipótese de ET, foram solicitados exames complementares para avaliar a presença de hamartomas em outros sistemas. Na USG de abdome realizado na primeira semana de vida não havia alterações. Porém no ano passado em consulta de seguimento foi solicitado novo exame de imagem que evidenciou presença de imagens sugestivas de angiolipomas renais. Atualmente a paciente está em seguimento com a Oncologia e Nefrologia.
Os nevos do tecido conjuntivo, são lesões extensas e em placas, de coloração normal ou marrom. Sendo chamadas de Manchas de Shagreen quando apresentam aspecto de casca de laranja, e estão localizados em região lombossacra. A placa fibrosa cefálica, é considerada a lesão mais específica da ET, e está presente em 25% dos pacientes. O fibroma ungueal ou Tumor de Koenen são tumores que aparecem mais tardiamente, após 2° década de vida, aumentam de tamanho progressivamente, mais comum em mulheres e acomete mais frequentemente os dedos dos pés. Na cavidade oral, as lesões mais frequentes são depressões no esmalte dos dentes e fibromas gengivais. (PORTOCARRERO LKL.,et al., 2018). O paciente pode apresentar lesões oculares, o hamartoma retinal acomete 30 a 50% dos pacientes, geralmente bilateral e múltipla, com início da infância. (PEREIRA C.C.S., et al.,2022)
Os guidelines atuais reafirmam a importância do diagnóstico genético, além do diagnóstico clínico. Podem ser identificadas variantes nos locus TSC1 e TSC2, principalmente, e a presença destas variantes são o suficiente para o diagnóstico, independente dos achados clínicos. O screening genético é uma alternativa para identificar os indivíduos com a doença muitas vezes antes dos sintomas se manifestarem. (NORTHRUP, H., et al., 2021). A ET é causada por variantes patogênicas nos genes TSC1 e TSC2, reconhecidos como genes supressores de tumores, localizados nos cromossomos 9q34.13 e 16p13.31, respectivamente. Várias variantes já foram sequenciados, porém novos sequenciamentos são necessários para que seja ampliado o banco genético relacionado à ET, uma vez que 10 a 15% dos pacientes diagnosticados clinicamente com ET não apresentam mutações sequenciadas. Devido a isso, as diretrizes internacionais afirmam que o teste genético sem alterações não exclui o diagnóstico de ET. (MONICH, A.G., et al., 2024)
Os critérios clínicos para diagnóstico da ET foram atualizados em 2021 nas diretrizes internacionais. Atualmente, são incluídos 11 critérios maiores e 7 critérios menores (Tabela 1). O diagnóstico definitivo é composto por 2 critérios maiores ou 1 maior associado a 2 menores. O diagnóstico possível é caracterizado por 1 critério maior ou pelo menos 2 menores. (NORTHRUP, H., et al., 2021)
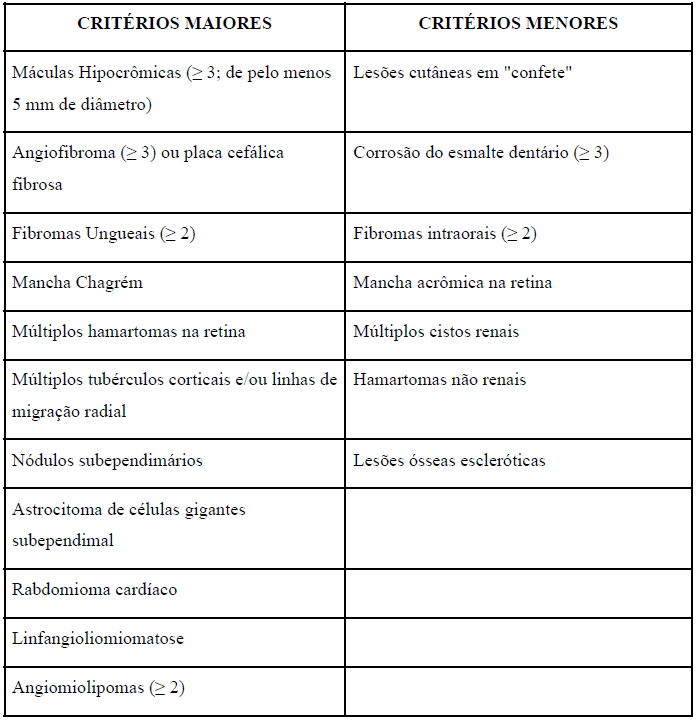
Tabela 01 – Critérios Clínicos Diagnósticos ET (DIRETRIZ – traduzido)
Como a ET é uma doença multissistêmica, após o diagnóstico ser realizado, o paciente deverá ser acompanhado por uma equipe multiprofissional. Do ponto de vista do acompanhamento genético, o ideal seria obter histórico de três gerações da família do paciente e avaliar o risco de ET nos familiares, a fim de garantir o melhor planejamento familiar, evitar complicações e possibilitar melhores prognósticos. (NORTHRUP, H., et al., 2021)
O ecocardiograma deve ser realizado em todos pacientes pediátricos, principalmente nos menores de 3 anos, para avaliação de rabdomiomas cardíacos. Devendo ser repetido anualmente em pacientes assintomáticos, até regressão do rabdomioma. O EEG deve ser realizado em todas as idades, para identificar erros na condução elétrica, e repetido a cada 3 a 5 anos, em pacientes assintomáticos. O ecocardiograma fetal é importante, uma vez que pode identificar o rabdomioma no pré-natal, a fim de evitar complicações ao nascimento. (NORTHRUP, H., et al., 2021)
A Ressonância (RNM) de Crânio, deve ser realizada em todos os pacientes com o diagnóstico, e caso não esteja disponível pode ser realizada Tomografia de Crânio ou USG fontanelar em neonatos. As diretrizes atuais recomendam que a RNM seja repetida a cada 1 a 3 anos, em assintomáticos e menores de 25 anos, para avaliar surgimento de astrocitoma. Nos pacientes que apresentam astrocitoma, a RNM deve ser realizada com maior frequência, para avaliar se há sinais de compressão, hidrocefalia ou outras complicações. (RIDDER, J.D., et al., 2021)
O eletroencefalograma (EEG) deve ser realizado durante o sono e vigília. (DAVID, P.E., et al., 2017). As crises epilépticas focais ou espasmos acontecem em 63 a 78% dos pacientes com ET, diante disso as diretrizes recomendam que o EEG seja realizado a cada 6 semanas, até paciente completar 1 ano de vida e após a cada 3 meses até completar 2 anos. Tal recomendação se baseia no fato de que a alteração no EEG acontece antes das repercussões clínicas. (NORTHRUP, H., et al., 2021)
Além dos exames citados acima, recomenda-se a realização de exame de imagem abdominal, independente da idade. A RNM de abdome é padrão-ouro para avaliar presença de angiolipomas, pois 30% são ricos em gorduras e podem não ser visualizados no USG de abdome. A RNM também pode avaliar a presença de aneurismas aórticos, hamartomas extra-renais e tumores neuroendócrinos do pâncreas. Quando não é possível realizar a RNM, pode ser feita a TC de abdome com contraste, para identificar cistos renais e angiomiolipomas pobres em gordura . A RNM de abdome deve ser repetida a cada 1 a 3 anos. Tais alterações renais podem resultar em uma hipertensão arterial secundária, portanto, os pacientes devem ter sua Pressão Arterial aferida em todas as consultas (KINGSWOOD, J. C., et al., 2020)
A avaliação clínica para linfangioleiomiomatose (LAM) deve ser realizada com Tomografia Computadorizada (TC) de tórax, em todas as mulheres e homens sintomáticos com 18 anos ou mais. Para as mulheres adultas com TC de tórax normal e que permaneceram assintomáticas, repetir exame a cada 5 a 7 anos até a menopausa. Nos pacientes em que for evidenciado doença cística no pulmão, é importante avaliar a função pulmonar do paciente com espirometria, os intervalos dos exames de acompanhamento devem ser determinados caso a caso. (TÆKKER, M., et al., 2021).
A consulta com o dentista deve ser realizada no momento da erupção do primeiro dente ou até 12 meses de idade e repetida a cada 6 meses. No caso do oftalmologista é importante a realização de fundoscopia em todos os pacientes com diagnóstico de ET, em busca de hamartomas ou lesões em retina. Os dermatologistas possuem importância significativa, uma vez que as lesões cutâneas são as mais prevalentes e por vezes permitindo até o diagnóstico precoce. As consultas devem ser anuais e ainda é recomendado o uso de protetor solar, uma vez que as máculas hipocrômicas são fotossensíveis. (NORTHRUP, H., et al., 2021)
Cerca de 90% dos paciente com ET apresentam manifestações neuropsiquiátricas. TAND (TSC-associated neuropsychiatric disorders) é uma termo que engloba todas essas manifestações da ET, entre elas as alterações comportamentais, psiquiátricas, intelectuais, acadêmicas, psicológicas e sociais. Portanto, também é importante que o paciente possua acompanhamento, para avaliação de todos os níveis das desordens psiquiátricas. Além disso, é necessário também instruir os pais e a família sobre os sinais de alarme de diversas desordens, como o TEA, déficit de atenção e hiperatividade, ansiedade e até dislexias. O médico assistente deve acompanhar o paciente e realizar testes validados de rastreamento para TAND, como o TAND Checklist, anualmente ou em menor período de tempo dependendo da necessidade do paciente. (MARCINKOWSKA, A.B, et al.,2022)
O tratamento da ET, consiste no manejo dos sintomas causados pelos hamartomas. Por se tratar de uma doença sistêmica, é obrigatório o acompanhamento multidisciplinar com a Genética, Neurologia, Neurocirurgia, Oftalmologia, Cardiologia, Pneumologia, Nefrologia, Dermatologia, Odontologia, Psiquiatria e Psicologia. (NORTHRUP, H., et al., 2021)
Em relação ao tratamento cirúrgico do astrocitoma de células gigantes subependimal deve ser recomendado nos pacientes com deterioração aguda do quadro clínico devido sangramento tumoral ou hidrocefalia causada pela obstrução do líquor. Atualmente, existem técnicas cirúrgicas minimamente invasivas como a ressecção endoscópica, mas esta é limitada a tumores pequenos e não é muito utilizada (FOHLEN, M., et al., 2018). Para o tratamento de tumores muito grandes ou irressecáveis com complicações, pode ser realizada uma derivação ventrículo peritoneal, para diminuir a pressão intracraniana e tratar a hidrocefalia. Além disso, pode ser realizado tratamento neoadjuvante com quimioterápicos, se as condições clínicas do paciente permitirem, assim, é possível diminuir a vascularização e o tamanho do tumor, possibilitando, talvez, a ressecção cirúrgica. (JANSEN, A. C., et al., 2019).
Alguns medicamentos podem ser utilizados nesses pacientes, como a vigabatrina, eficaz nas convulsões focais e espasmos infantis, principalmente em portadores de ET.
Quanto mais precoce é iniciado, melhor é a resposta nas crises refratárias. Porém os efeitos adversos relacionados à retinopatia resultam em defeitos permanentes do campo visual periférico, o que têm limitado a sua aplicação na ET. (GOLEC,W., et al., 2021)
Após a descoberta da regulação da via mTOR e sua relação com desenvolvimento de tumores, alguns estudos promissores têm sido feitos favorecendo a possibilidade de tratamento de pacientes com ET de acordo com fisiopatologia da doença. O primeiro inibidor do mTOR a ser descoberto foi a rapamicina. Porém, devido às características farmacocinéticas e farmacodinâmicas, a sua toxicidade, instabilidade e baixa biodisponibilidade, foi necessário desenvolver análogas da rapamicina. (ARONICA, E., et al, 2023)
O Everolimus é um inibidor da mTOR que tem sido estudado em pacientes com ET desde 2006, com boa resposta no tratamento dos angiolipomas renais, astrocitomas subependimários de células gigantes e linfangioleiomiomatose, com benefícios secundários nas manifestações cutâneas. (PAOLO, C., et al., 2022). Alguns estudos também demonstram bons resultados no tratamento para epilepsia refratárias na ET, no entanto, ainda não há consenso na literatura ou recomendação formal. (MENEZES, C.E.G., et al, 2023) E apesar de ser um medicamento de alto custo é seguro e eficiente, principalmente quando se trata de sintomas neurológicos. (ARONICA, E., et al, 2023).
No que diz respeito aos efeitos cutâneos dos inibidores de mTOR, alguns estudos têm demonstrado a resposta do Sirolimus às lesões como: angiofibroma facial, fibromas ungueais e manchas Shagreen. Porém as novas diretrizes limitam o uso de inibidores orais de mTOR para lesões cutâneas em pacientes não elegíveis para abordagens cirúrgicas e cujas lesões apresentem risco de hemorragias extensas e recorrentes, uma vez que o tratamento oral está associado a risco aumentado de infecções como estomatite, amenorreia, acne e alterações laboratoriais. (MENEZES, C.E.G., et al, 2023)
Os angiofibromas faciais surgem geralmente na infância e estão associados ao risco de sangramento e por vezes a questões psicológicas devido à aparência. O tratamento padrão é cirúrgico, como ablação por radiofrequência, crioterapia, eletrocoagulação, dermoabrasão, laser cirurgia e aplicação de podofilotoxina. Alguns destes são caros, enquanto outros são destrutivos, podem levar à formação de cicatrizes e tem seus riscos associados à anestesia e recuperação pós-cirúrgica. Diante disso o tratamento tópico tem sido sugerido como abordagem segura e eficaz para o tratamento do angiofibroma. O Sirolimus tópico 0,2% gel foi considerado eficaz e com boa tolerância no tratamento de lesões cutâneas, principalmente o angiofibroma facial, com taxa de resposta superior a 80%. (EGAMI, A., et al, 2023)
Por fim, a ET é uma doença que não possui cura e, dessa forma, é muito importante acompanhar o paciente durante toda a vida, por meio de exames de rotina e acompanhamento do quadro clínico com equipe multiprofissional. E quando necessário intervir nas complicações da doença e oferecer a melhor qualidade de vida possível para o paciente.
CONCLUSÃO
A ET pode ser uma doença devastadora, com grande morbidade na vida do paciente. E por se tratar de uma doença multissistêmica o paciente precisa realizar exames complementares com o objetivo de rastrear tumores em todo o organismo. O ecocardiograma, RNM de crânio, RNM de abdome e TC de tórax são exemplos de exame de imagem que precisam ser realizados de rotina, além de um acompanhamento multiprofissional do paciente, principalmente durante a infância e adolescência, período em que há maior probabilidade do início das tumorações. Dessa maneira, é possível diagnosticar precocemente as tumorações e complicações da doença. Os guidelines atuais reafirmam também a importância dos teste genéticos, os quais são capazes também de antecipar diagnósticos, além de promover aconselhamento genéticos para os familiares. Conclui-se portanto que o diagnóstico precoce, o acompanhamento multiprofissional e testes genéticos são fundamentais para melhor prognóstico do paciente, melhorando a qualidade vida e sobrevida destes pacientes.
REFERÊNCIAS BIBLIOGRÁFICAS
ARONICA, E., et al. Epileptogenesis in tuberous sclerosis complex-related developmental and epileptic encephalopathy. Brain, v. 146, n. 7, p. 2694-2710, 2023.
DAVID, P. E., et al. Presentation and diagnosis of tuberous sclerosis complex in infants. Pediatrics, v. 140, p. e20164040, 2017.
EGAMI, A., et al. Topical Sirolimus 0.2% Gel for the Management of Tuberous Sclerosis Complex-Related Cutaneous Manifestations: An Interim Analysis of Postmarketing Surveillance in Japan. Dermatol Ther (Heidelb), v.13, p. 1113-1126, 2023.
FOHLEN, M., et al. Surgery for subependymal giant cell astrocytomas in children with tuberous sclerosis complex. Childs Nerv Syst, v. 34, p. 1511-1519, 2018.
GOLEC,W., et al. Vigabatrin — new data on indications and safety in paediatric epilepsy. Polish Journal of Neurology and Neurosurgery, v.55, n.5, p 429-439, 2021
JANSEN, A. C., et al. Clinical characteristics of subependymal giant cell astrocytoma in tuberous sclerosis complex. Front Neurol, v. 10, p. 705, 2019.
KINGSWOOD, J. C., et al. Renal manifestations of tuberous sclerosis complex: key findings from the final analysis of the TOSCA study focussing mainly on renal angiomyolipomas, Front Neurol, v. 11, p. 972, 2020.
MARCINKOWSKA, A.B, et al. Tuberous Sclerosis Complex Patients’ Needs and Difficulties—Results of TAND Questionnaire Analysis in Polish Adult Population. Journal of Clinical Medicine, v.11, p.6536, 2022
MENEZES, C.E.G., et al. Everolimus as a therapeutic option in refractory epilepsy in children with tuberous sclerosis:a systematic review. Academia Brasileira de Neurologia, v. 81, p. 392-398, 2023.
MONICH, A.G., Bissler J.J., Barreto, F,C. Tuberous Sclerosis Complex and the kidneys: what nephrologists need to know.Brazilian Journal of Nephrology, v.46(3):e20240013, 2024.
NORTHRUP, H., et al. Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatric Neurology, v. 123, p. 50-66, 2021.
PAOLO, C., et al. Advances in the genetics and neuropathology of tuberous sclerosis complex: edging closer to targeted therapy. The Lancet Neurology, v. 21, n. 9, p. 843-856, 2022.
PEREIRA CCS., et al. Clinical profile of tuberous sclerosis complex patients with and without epilepsy: a need for awareness for early diagnosis. Arquivos neuropsiquiatria. 2022; 80 (10):1004-1010
PORTOCARRERO LKL.,et al. Tuberous sclerosis complex: review based on new diagnostic criteria. Anais Brasileiros de Dermatologia. 2018;93(3):323
RIDDER, J.D., et al. Early epileptiform EEG activity in infants with tuberous sclerosis complex predicts epilepsy and neurodevelopmental outcomes. Epilepsia, v. 62, p. 1208-1219, 2021.
TÆKKER, M., et al. Diagnostic accuracy of low-dose and ultra-low-dose CT in detection of chest pathology: a systematic review, Clinical Imaging, v. 74, p. 139-148, 2021.
1Graduada em Medicina pela Universidade Católica de Brasília (UCB). QNC,E-mail: julianafaleirop@gmail.com;
2Graduada em Medicina pela Universidade de Brasília (UnB) Campus Universitário Darcy Ribeiro, E-mail: giribeirosil@gmail.com;
3Graduada em Medicina pela Universidade Católica de Brasília (UCB). QNC, E-mail: ekaterine_apostolos@hotmail.com;
4Graduado em Medicina pelo Centro Universitário de Brasília (UNICEUB). Faculdade de Ciências da Educação, E-mail: eduardojfsales@gmail.com;
5Graduada em Medicina pelo Centro Universitário do Planalto Central Apparecido dos Santos (UNICEPLAC). SIGA E-mail: anacarolbuta@gmail.com;
6Graduada em Medicina pela Universidade Federal de Goias (UFG). E-mail: bastosheloise1@gmail.com;
7Graduada em Medicina pelo Centro Universitário do Planalto Central Apparecido dos Santos (UNICEPLAC). SIGA E-mail: coutolimaana@gmail.com;
8Graduada em Medicina pela Universidade Católica de Brasília (UCB). E-mail: vit_fulanette@hotmail.com;
9Graduado em Medicina pelo Centro Universitário de Brasília (UNICEUB). Faculdade de Ciências da Educação, E-mail: renatamachadobonfim@gmail.com;
10Especialista em Cardiologia Pediátrica e Ecocardiografia Pediátrica pelo Instituto de Cardiologia e Transplantes do Distrito Federal. E-mail: lucianycarvalho@gmail.com