PHARMACEUTICAL EQUIVALENCE AND BIOEQUIVALENCE: PROOFING INTERCHANGEABILITY BETWEEN MEDICINES, ENABLING ACCESS TO HEALTHCARE
REGISTRO DOI: 10.5281/zenodo.10204159
Jaqueline Terras Mendes1
Lucimar de Melo Nogueira1
Orientadora: Profª Mestra Carin Fabíola Pensin2
Coorientadora: Profª Mestra Patrícia Moura da Rosa Zimmermann2
Resumo
As formulações farmacêuticas são cada vez mais utilizadas para a prevenção, restauração e manutenção da saúde, por possibilitarem a melhora da qualidade de vida aos indivíduos. Contudo, o alto custo no tratamento medicamentoso dificulta a continuidade do tratamento para a população mais carente. Para ampliar esse acesso, o Brasil vem implementando políticas que propiciam o acesso à saúde. Esse movimento teve início em 1998, com a lei nº 3.916 sancionada pelo governo federal, que constituiu a Política Nacional de Saúde (PNM) como fundamento em prol da melhoria contínua da saúde da população. O avanço para a acessibilidade a tratamento de qualidade através da comercialização dos medicamentos genéricos, obteve grande impacto em 1999 com o surgimento da Lei do genérico com estabelecimento da política de comercialização de medicamentos genéricos. Esta teve o intuito de garantir que a parcela mais carente da população tivesse acesso ao tratamento gratuito ou ainda conseguisse adquirir medicamentos por meio de preços efetivamente mais baixos, com a qualidade, segurança e eficiência terapêutica comprovadas. Nesse cenário, os estudos de equivalência farmacêutica e bioequivalência farmacêutica, dentre outras exigências de boas práticas, são os principais requisitos no processo regulatório para obtenção do registro de medicamentos genéricos no Brasil. Estes precisam ser aprovados em atendimento a requisitos de qualidade, sendo intercambiáveis ao medicamento de referência, possuindo, portanto, equivalência terapêutica. Assim, o objetivo desse trabalho é abordar as principais premissas necessárias em legislação, literatura científica e compêndios oficiais, acerca da qualidade dos medicamentos genéricos comercializados no Brasil, com ênfase em resultados de estudos de Equivalência e Bioequivalência Farmacêutica realizados para o registro de medicamentos genéricos.
Palavras-chave: Intercambialidade. Medicamentos Genéricos. Equivalência Farmacêutica. Bioequivalência Farmacêutica.
1. INTRODUÇÃO
A busca pela cura de doenças e alívio das dores é um objetivo constante da humanidade desde os tempos antigos. Registros históricos mostraram que o uso de plantas, animais e minerais para o tratamento de doenças existiam antes mesmo de Cristo. A ampliação da extração de princípios ativos ocorreu somente por volta do século XX. Com o avanço dos tempos e a evolução da ciência e da medicina, surgiram as primeiras pesquisas para o desenvolvimento dos primeiros fármacos, até chegar-se ao medicamento conhecido nos dias atuais (CALIXTO e SIQUEIRA JUNIOR, 2008).
No Brasil, nesse período, a indústria farmacêutica já se destacava com a produção de soros e vacinas. Isso aconteceu nas cidades de Manguinhos (RJ) e no Instituto Bacteriológico de São Paulo (PINTO E BARREIRO, 2013).
O processo de desenvolvimento de novas moléculas com potencial à novos fármacos é um trabalho multidisciplinar que envolve médicos, químicos, engenheiros, biólogos, farmacêuticos e outros profissionais. Com isso, as indústrias farmacêuticas realizam diversas parcerias com universidades em busca de acelerar as pesquisas e possibilitar maior disponibilidade de tratamento em médio e curto prazo à população (CALIXTO e SIQUEIRA JUNIOR, 2008).
O medicamento é considerado o produto de cunho farmacêutico contendo no mínimo um fármaco (princípio ativo), elaborado para a finalidade curativa, profilática ou para o diagnóstico. Ainda, pode conter em sua formulação associações de fármacos, adjuvantes farmacoterapêuticos e excipientes. O medicamento também deve apresentar segurança, qualidade, eficácia terapêutica comprovadas por meio de análises criteriosas (ALVES et al., 2012).
Em 1960 nos Estados Unidos da América, iniciaram as ações favoráveis à industrialização dos medicamentos genéricos. O intuito era ampliar a disponibilidade de tratamento para as classes econômicas de menor poder aquisitivo (SILVA et al., 2022).
O alto custo de muitos dos medicamentos disponíveis dificultam o acesso ao tratamento à população de baixa renda, impedindo-os de iniciar ou continuar o tratamento de forma adequada (BRUNNA et., al, 2022). No Brasil, nesse mesmo período, já era necessária a adoção de tratativas aos problemas de saúde pública existentes em função da escassa oferta de medicamentos à preços acessíveis (SILVA et al., 2022).
Demorou-se um pouco mais até se pensarem em medidas para o atendimento às necessidades voltadas à classe social mais carente. Contudo, com o passar dos anos, essa perspectiva foi gradativamente alcançada (SILVA et al., 2022).
Figura 01: Linha do tempo do cenário regulatório do medicamento no Brasil.
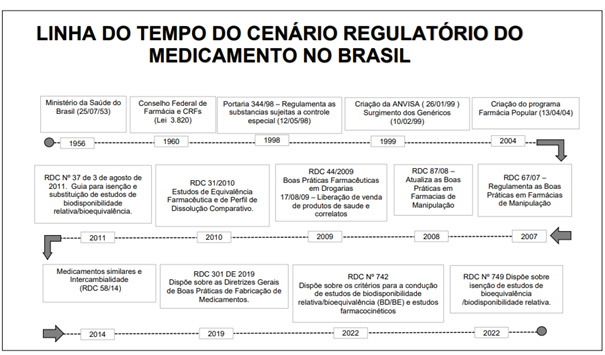
Fonte: Adaptado pelos autores, 2023.
Em 1999 foi sancionada a Lei Nº 9.787, lei dos genéricos e instituiu a utilização dos medicamentos genéricos no Brasil que tem como objetivo propiciar a maior disponibilidade de tratamento, alavancar a economia e diminuir os preços dos medicamentos de referência, a lei dos genéricos complementou a Política Nacional de Saúde (PNM), fundamentada em promover a melhoria contínua das condições de saúde da população (BRASIL, 2001).
Com o surgimento do mercado de medicamentos genéricos foram necessárias adequações relevantes de cunho regulatório. Essas adequações foram essenciais para organizar as vias aos interessados em adentrar nesse cenário de mercado. Padronizações, conceitos, legislações e portarias foram imputadas pelo governo federal e por órgãos reguladores competentes. Assim, as indústrias farmacêuticas tiveram que estabelecer medidas e prazos para as adequações necessárias ao atendimento dos requisitos de qualidade exigidos pela Agência Nacional de Vigilância Sanitária (ANVISA). As modificações também foram propostas para elevar o respaldo aos consumidores dos medicamentos genéricos (LEMES, et al, 2018).
Para que um medicamento seja registrado como genérico é imprescindível, segundo a ANVISA, a comprovação da sua equivalência terapêutica. Comprovada por meio de ensaios in vitro e in vivo que avaliam a comparabilidade de uma formulação teste, candidata a genérico, frente a formulação de referência, tal comprovação é fundamental para a sustentação da intercambialidade entre os medicamentos (BRASIL, 2004).
O medicamento de referência é um produto inovador registrado no órgão federal responsável pela vigilância sanitária e comercializado no Brasil, cuja eficácia, segurança e qualidade foram comprovadas cientificamente junto ao órgão federal competente, por ocasião do registro, portanto, passou por ensaios clínicos que demonstram sua eficácia e segurança em pacientes. O medicamento genérico possui a comprovação terapêutica quando avaliado comparativamente ao medicamento de referência, nos ensaios de equivalência farmacêutica. Possui também a seguridade terapêutica, por meio da avaliação clínica, analítica e estatística de desempenho quando aplicado em estudos de bioequivalência/ biodisponibilidade farmacêutica, em dose equivalente ao medicamento de referência, indicado pela ANVISA. O medicamento genérico possui as mesmas características: forma farmacêutica, dosagem, concentração e demais aspectos em igualdade ao medicamento de referência, e deve apresentar semelhante confiabilidade, atribuindo-lhe, nesse caso, o título de intercambiável ao de referência, podendo substituir o mesmo, sem impacto clínico (LEMES, et al, 2018).
Consideram-se medicamentos equivalentes farmacêuticos, os quais cumprem fielmente e igualmente as exigências da Farmacopeia brasileira, ou de outras monografias desde que justificada tecnicamente a utilização de monografia diferente. Os medicamentos submetidos ao teste de equivalência, devem ter a mesma via de administração, em mesma dosagem da substância ativa (mesmo sal ou éster) e podem ou não conter os excipientes idênticos desde que delineados para a condição desejada (ANVISA, 2020).
No estudo de equivalência farmacêutica algumas particularidades fundamentais devem ser atendidas nas análises dos medicamentos testes e de referência em mesma metodologia de análise: pH, umidade, doseamento (determinação de teor e uniformidade de doses unitárias), quantificação de substâncias químicas (impurezas e substâncias relacionadas), quantificação da velocidade da dissolução (ensaio de dissolução e perfil de dissolução) e ensaios de identificação do fármaco (ANVISA, 2020).
O objetivo do estudo de Bioequivalência Farmacêutica, segundo a Resolução 1.170 de 2006 (RE 1.170, DE 19 DE ABRIL DE 2006[CdM1] ) é determinar a bioequivalência de um medicamento genérico em comparação aos resultados do medicamento de referência. O estudo é realizado por meio da quantificação do fármaco e/ou do metabólito ativo na circulação (sangue, plasma ou soro) ou
por meio da sua quantificação na urina, quando justificado. Alternativamente, também poderá ser realizado comparando medidas farmacodinâmicas. Nesse sentido, avalia-se e compare-se a taxa de absorção e quando semelhante, comprova-se a similaridade entre os medicamentos, garantindo a qualidade, eficácia terapêutica e segurança do medicamento candidato a genérico (RE nº1.170, 2006).
Ainda segundo a RE 1.170,2006 as etapas gerais do estudo de bioequivalência, são: etapa clínica: planejamento, protocolo, seleção dos voluntários, administração dos medicamentos e coleta dos dados (coletas sanguíneas); etapa Analítica: interpretação dos dados coletados e análise farmacocinética; etapa estatística: Análises estatísticas e relatório dos resultados, submissão regulatória e decisão regulatória. É importante observar que as diretrizes e os requisitos regulatórios específicos para estudos de bioequivalência podem variar de um país para outro. Portanto, os estudos de bioequivalência devem ser elaborados de acordo com as regulamentações e diretrizes locais. Além disso, a segurança dos participantes é de extrema importância, e os estudos são conduzidos com prazos e padrões rigorosos, éticos e com segurança (RE nº1170, 2006).
Nesse sentido, serão abordados os requisitos necessários em legislação, literatura e compêndios oficiais que asseguram a qualidade, segurança e a eficácia terapêutica do medicamento genérico registrado e comercializado no Brasil. As pesquisas bibliográficas e aprofundamentos relevantes sobre o tema em questão, apresentarão conteúdos pautados na legislação vigente, formalizando as etapas, conceitos e parâmetros utilizados nos estudos de equivalência e bioequivalência farmacêutica. E possuem como o objetivo, explanar os passos relevantes para um estudo de Equivalência e Bioequivalência que visam garantir a eficácia, a qualidade e a segurança para a população em tratamento medicamentoso.
2. JUSTIFICATIVA
A revisão bibliográfica que se pretende realizar, prevê a apresentação dos dados de um estudo de equivalência e bioequivalência farmacêutica realizados para o registro de um novo medicamento genérico. Em razão da estratégia regulatória que o medicamento candidato a genérico requer para a obtenção de autorização de registro previamente a comercialização, justifica-se o interesse pela abordagem acadêmica acerca do assunto.
Portanto, com a conclusão desse trabalho se almeja a contribuição para o entendimento dos critérios pelos quais os medicamentos genéricos são previamente submetidos, os quais asseguram a qualidade, eficácia terapêutica e segurança, permitindo a intercambialidade com o medicamento de referência[CFP2] .
2.1. OBJETIVOS GERAIS
Abordar as principais premissas necessárias em legislação, literaturas e compêndios oficiais, acerca da qualidade dos medicamentos genéricos comercializados no Brasil, com ênfase em resultados de estudos de Equivalência e Bioequivalência Farmacêutica realizados para o registro de um medicamento genérico.
2.2. OBJETIVOS ESPECÍFICOS
- Demonstrar os testes em que são submetidos os medicamentos candidatos a genéricos no Brasil;
- Ampliar o conteúdo disponível sobre a qualidade, segurança e eficácia terapêutica dos medicamentos genéricos;
- Apresentar os conceitos relevantes acerca dos estudos de equivalência e bioequivalência farmacêutica, evidenciando a importância destes, na garantia da qualidade e segurança, bem como se aplica o conceito de intercambialidade.
2.3. QUESTÕES NORTEADORAS
- O que é o estudo de Equivalência e Bioequivalência Farmacêutica?
- Como são conduzidos esses estudos?
- Como é realizada a avaliação do medicamento previamente a sua comercialização?
- O que são os medicamentos intercambiáveis?
3. FUNDAMENTAÇÃO TEÓRICA OU REVISÃO DA LITERATURA
A humanidade desde o início dos tempos evidencia uma constante busca para a cura das dores e doenças. Os primeiros registros do uso de plantas, animais e minerais para o tratamento de doenças, foram datados antes mesmo da existência de Cristo. Esses dados antigos deixados por diferentes civilizações, apontam que a extração de ativos de plantas medicinais para o uso farmacoterapêutico iniciaram por volta do século XIX. Muito tempo depois surgiram as primeiras pesquisas para o desenvolvimento de novos fármacos, os primeiros princípios ativos e formas farmacêuticas, dentre outras características, até chegar-se a diversidade de medicamentos presentes no início da terapêutica (CALIXTO e SIQUEIRA JUNIOR, 2008).
Por volta do século XX, nascia a indústria farmacêutica moderna que evoluiu[CFP3] [LN4] constantemente até chegar ao patamar atual (CALIXTO e SIQUEIRA JUNIOR, 2008). O desenvolvimento de uma molécula com potencial a novo fármaco é sem dúvidas um trabalho para equipes multidisciplinares: médicos, químicos, engenheiros, biólogos, farmacêuticos, dentre outros, desempenham esforços relevantes em pesquisas para elevar o portfólio das indústrias farmacêuticas.
Após o acidente trágico acontecido nos Estados Unidos, por volta de 1938, em que 76 pessoas perderam suas vidas ao serem intoxicadas pela ingestão de um medicamento contendo 72% de dietilglicerol empregado na formulação da Sulfonamida como solvente, evidenciou-se a necessidade de estudos clínicos prévios ao uso de medicamentos em humanos. Desde então foi estabelecida a participação de voluntários nos estudos clínicos, além de critérios básicos que os medicamentos deveriam apresentar antes da administração a humanos. Em razão do acontecimento surgiram os ensaios clínicos, requeridos até hoje: de fase I, II e III necessários para a avaliação da segurança e eficácia de um novo medicamento (CALIXTO e SIQUEIRA JUNIOR, 2008). Vinculada ao estado, na maioria das vezes, a indústria farmacêutica brasileira apresentava nesse período notáveis avanços no desenvolvimento de fármacos. Um dos exemplos foi a produção de soros e vacinas nas cidades de Manguinhos (RJ) e no Instituto Bacteriológico de São Paulo, no início do século XX (PINTO E BARREIRO, 2013).
A Agência Nacional de Vigilância Sanitária (ANVISA 2010) define que medicamentos são produtos de cunho farmacêutico especialmente elaborados para o diagnóstico, prevenção, cura ou alívio de sintomas e doenças. Atualmente sua produção exige rigoroso sistema de gerenciamento da qualidade em todas as etapas produtivas. Acompanhadas e certificadas por esse órgão regulador, as exigências visam a seguridade ao paciente no decorrer do tratamento. O efeito de um medicamento está relacionado a sua substância ativa a qual produz efeitos terapêuticos conhecidos cientificamente.
Vinculada ao estado, na maioria das vezes, a indústria farmacêutica brasileira apresentava nesse período notáveis avanços no desenvolvimento de fármacos. Um dos exemplos foi a produção de soros e vacinas nas cidades de Manguinhos (RJ) e no Instituto Bacteriológico de São Paulo, no início do século XX (PINTO E BARREIRO, 2013).
O uso de um medicamento está relacionado aos conceitos de saúde e doença independendo da classe social, cultura ou história do indivíduo. É considerado um medicamento toda a preparação de cunho farmacêutico adequada ao uso, na qual foi adicionado um fármaco ou uma associação de fármacos (princípio ativo). Ainda, poderão conter nessa preparação além do fármaco, adjuvantes farmacêuticos e excipientes. O medicamento é produzido para o consumo humano com a finalidade curativa, profilática ou de diagnóstico (ALVES et al., 2012).
O fármaco é o princípio ativo em uma formulação farmacêutica. É o componente que apresentará o efeito farmacológico ativo em um medicamento. Esse efeito farmacológico é conhecido, certificado, qualificado e possui a validação metodológica a comprovada. Assim, a forma farmacêutica final contendo o fármaco deve apresentar por obrigatoriedade os registros que permitam a rastreabilidade de todo o processo. Esses controles rigorosos desempenham sobre a formulação farmacêutica a capacidade de assegurar a qualidade, eficácia terapêutica e a segurança ao serem administrados à humanos (ANVISA, 2007).
ANVISA destaca que a produção do medicamento segue normas rígidas assistidas desde a sua pesquisa, desenvolvimento aos testes de qualidade e posteriormente, na comercialização, salienta ainda que o uso do medicamento deve ser preconizado e orientado pelos profissionais da saúde responsáveis (ANVISA 2010). Atualmente a maioria dos compostos químicos destinados à fármacos são de origem natural ou obtidos pela síntese de produtos naturais. Plantas, fungos, insetos, organismos marinhos e bactérias são fontes primordiais para a extração de moléculas bioativas. O planejamento racional de fármacos foi delineado por Emil Fischer em 1885 no modelo de chave-fechadura, o qual reforça a teoria dos receptores (sítios de ligação da molécula terapêutica) colaborando diretamente para o desenvolvimento da química medicinal (NASCIUTTI, 2012)
3.1 MEDICAMENTO GENÉRICO: INTERCAMBIALIDADE MEDICAMENTOSA COM SEGURANÇA, QUALIDADE E EFICIÊNCIA TERAPÊUTICA.
Por volta de 1960, nos Estados Unidos já se iniciavam as ações para a industrialização do medicamento genérico com o intuito de alavancar a economia e melhorar o acesso ao tratamento medicamentoso às classes econômicas com menor poder aquisitivo. O Brasil, espelhando-se a esse e à demais países do Ocidente, demonstrou também o interesse pela comercialização dos medicamentos genéricos, como tratativa a um problema de saúde pública que se emanava. Em 1993, surgiu a Lei dos medicamentos genéricos, que configurava a PNM (Política Nacional de Medicamentos) em prol da qualidade de vida da população mais carente (SILVA et al., 2022).
Porém, somente em 1999 a política brasileira regulamentadora dos medicamentos genéricos foi instituída com a Lei nº 9.787. A regulação da Lei dos genéricos ocorreu em função da Resolução da Diretoria Colegiada da Agência Nacional de Vigilância Sanitária (ANVISA), a qual evidenciou os critérios mínimos obrigatórios para o registro e comercialização de medicamento genérico no Brasil (ARAÚJO et al., 2010).
Houve a partir de então a formação de diferentes legislações com a finalidade de assegurar a padronização da produção dos medicamentos genéricos, os quais por determinação da ANVISA, devem possuir a comprovação da eficácia terapêutica, segurança e qualidade. Sendo que os medicamentos genéricos devem obedecer aos mesmos requisitos que o medicamento de referência indicado pela ANVISA possui (LEMES, et al., 2018).
Certamente, a Lei dos genéricos, foi um grande estímulo para o desenvolvimento da Indústria Farmacêutica Nacional. A partir de então observou-se demasiado e crescente incentivo financeiro, além do fiscal, os quais contribuíram para o surgimento de diversas parcerias multinacionais, dedicadas em grande parte ao fornecimento dessa classe de medicamentos de forma ampliada (CALIXTO e SIQUEIRA JUNIOR, 2008).
Para a Organização Mundial da Saúde (OMS) o medicamento genérico é o produto farmacêutico intercambiável com o produto inovador. Geralmente desenvolvido ainda com a patente do medicamento inovador válida, porém comercializado após a expiração da proteção patentearia (BERMUDEZ, 1994).
A ANVISA determina que o medicamento genérico é o produto farmacêutico que contenha o mesmo princípio ativo, na mesma dose e forma farmacêutica, e administrado pela mesma via de administração. Deve apresentar também a mesma posologia e indicação terapêutica do medicamento de referência, contendo eficácia e segurança equivalentes a este. As embalagens dos medicamentos genéricos devem trazer a informação do princípio ativo, possuir uma tarja amarela com a escrita “Medicamento Genérico” e ainda, obrigatoriamente conter a frase “Medicamento Genérico Lei nº 9.787/99” (ANVISA 2010).
Figura 1: Características da embalagem do medicamento genérico

FONTE: Fundação Oswaldo Cruz, 2007.
A intercambialidade é o termo oficial que justifica a possibilidade da substituição do medicamento de referência pelo genérico, mediante a autorização médica, necessidade ou escolha do paciente. Segundo o RDC Nº 16, DE 2 DE MARÇO DE 2007 dois medicamentos são intercambiáveis quando são equivalentes terapêuticos, comprovando essencialmente, os mesmos efeitos de eficácia e segurança. A intercambialidade é assegurada, quando após a administração na mesma dose molar, ambos medicamentos apresentam os mesmos efeitos avaliados em ensaios apropriados de bioequivalência farmacêutica (ANVISA, 2010).
Todo o medicamento candidato a genérico ao ser submetido aos testes de desempenho in vitro ou in vivo deve ter como o medicamento intercambiável o de referência listado em documento regulamentado pela ANVISA (DE OLIVEIRA, et al, 2023).
Serão formulações equivalentes, ao conter o mesmo fármaco (mesmo sal ou éster da mesma molécula terapeuticamente ativa), em quantidades e formas farmacêuticas idênticas. Ainda, adicionalmente, quando manter as especificações mínimas requeridas na Farmacopeia Brasileira ou em outros compêndios, desde que autorizados. A substituição do medicamento genérico pelo de referência é assegurada pelos resultados dos ensaios da equivalência terapêutica, que incluem comparação in vitro, e estudos de bioequivalência farmacêutica in vivo, apresentados à Agência Nacional de Vigilância Sanitária (ANVISA, 2010).
3.2. ESTUDOS DE EQUIVALÊNCIA FARMACÊUTICA
De acordo com a RDC 31/2010 (RESOLUÇÃO-RDC Nº 31, DE 11 DE AGOSTO DE 2010) os estudos de equivalência são ensaios físico-químicos, e quando aplicáveis microbiológicos e biológicos, que possuem a finalidade da certificação da equivalência farmacêutica entre dois medicamentos. Esses ensaios físico-químicos devem ser realizados por centros de estudos devidamente habilitados e regulados pela ANVISA. Os estabelecimentos que conduzem os estudos de equivalência farmacêutica são juridicamente responsáveis em conjunto com a empresa patrocinadora do estudo, pelos dados técnicos registrados em todo o decorrer dos testes até a finalização e entrega de resultados e relatórios (ANVISA, 2010).
Os testes físico-químicos aplicados no estudo de equivalência farmacêutica, visam a avaliação do comportamento farmacocinético e farmacodinâmico de ambas formulações em análise (medicamento teste e de referência) para a certificação da semelhança entre elas. São ensaios in vitro determinantes para a intercambialidade entre o candidato a genérico e o de referência (DE OLIVEIRA, et al, 2023).
Os medicamentos que são analisados para a certificação de equivalentes farmacêuticos devem estar dentro do prazo de validade e acondicionados de forma apropriada. O medicamento teste juntamente com os insumos e materiais analíticos, devem ser enviados ao centro de equivalência pelo patrocinador do estudo, enquanto que o medicamento de referência é considerado lote comercializado e pode ser adquirido direto do mercado, pelo centro de estudos. Ambos os medicamentos de referência e teste devem cumprir com os requisitos da monografia individual, preferencialmente com a farmacopeia Brasileira ou baseados em demais compêndios oficiais, ou em normas e regulamentos específicos referendados pela ANVISA (ANVISA, 2010).
Quando na ausência de monografias ou compêndios oficiais, deve ser utilizado metodologia analítica validada pelo patrocinador do estudo ou centro de equivalência farmacêutica, salienta-se que ambos medicamentos devem apresentar compatibilidade e resultados de acordo com o previsto pelo método analítico determinado. Ainda sobre os medicamentos empregados nos estudos de equivalência farmacêutica, é importante relatar que os mesmos lotes de medicamentos testes e de referência avaliados nesses ensaios devem ser conduzidos, quando aplicável, aos estudos de bioequivalência farmacêutica (ANVISA, 2010).
Para a realização de tais estudos, a ANVISA, órgão regulamentador competente, designou a listagem dos laboratórios com petições atendidas no que tange as normas e especificações necessárias requeridas na RDC 67 de 2016 (RDC Nº 67, DE 23 DE MARÇO DE 2016), que dispõe sobre os critérios para as petições aos registros dos Centros de Equivalência Farmacêutica. Além disso, é válido acrescentar sobre a existência da certificação REBLAS (Rede Brasileira de Laboratórios Analíticos em Saúde), que fortalece a confiabilidade dos resultados apresentados. Possuir a certificação de laboratório REBLAS, imputa ao laboratório ser condizente, capaz e habilitado a conduzir os serviços de interesse sanitário, pautados na confiabilidade, qualidade, segurança e em condições sistematicamente rastreáveis (ANVISA, 2020).
Também é possibilitado às indústrias farmacêuticas desenvolverem dentro de seus espaços físicos seus próprios centros de equivalência farmacêutica. Para elas o investimento é válido pois correspondem a diminuição dos prazos para a obtenção dos registros (DE OLIVEIRA, et al, 2023).
Quanto aos ensaios necessários para a certificação de equivalência farmacêutica, a ANVISA determina que alguns destes são obrigatórios e outros apenas informativos, no entanto, os informativos podem ser considerados também de obrigatoriedade quando seus resultados sejam necessários para validar a qualidade terapêutica do fármaco em questão. Dentre os ensaios obrigatórios do estudo de equivalência farmacêutica, está o ensaio de Dissolução. O ensaio de dissolução do produto também precisa ser pautado individualmente em compêndios oficiais, farmacopéia brasileira ou normas específicas referendadas pela ANVISA. Em exceção, para os casos em que não seja utilizada a metodologia oficial o patrocinador do estudo deve apresentar metodologia de dissolução desenvolvida com base nos guias nacionais e internacionais comprovando que o método local é discriminativo (ANVISA, 2010).
O centro de estudos responsável pela execução do serviço deve arquivar cópia do relatório de desenvolvimento de dissolução fornecida pela empresa patrocinadora. Tal relatório precisará atender alguns critérios mínimos, tais como: avaliar quantitativamente a solubilidade do ativo em diferentes faixas de pH fisiológico que podem variar de 1,2 a 6,8 pH em temperatura de 37°C ± 1°C; demonstrar por meio da avaliação da curva da dissolução nos diferentes meios de pH fisiológicos que o meio de dissolução empregado é o apropriado para a forma farmacêutica em análise, assim como, os aparatos, rotação e filtros escolhidos para a avaliação das amostras, dentre outros fatores (ANVISA, 2010).
No estudo de equivalência Farmacêutica deve-se avaliar o perfil de dissolução comparativo entre o fármaco teste e o de referência. O estudo de perfil de dissolução comparativo (PDC) é executado nos mesmos critérios do estudo de dissolução realizado no estudo de equivalência farmacêutica. Os medicamentos a serem analisados devem ser os mesmos lotes do estudo de equivalência e Bioequivalência farmacêutica, quando aplicável. Esse ensaio tem por finalidade, a avaliação da dissolução da substância ativa em vários pontos de coletas, diferente do ensaio de dissolução que avalia apenas um ponto de coleta. Os dados coletados são avaliados comparando-se os resultados de ambas formulações (ANVISA, 2010). A razão dos resultados do teor dos medicamentos teste e de referência não deve ser superior a 5% (BRASIL, 2003).
Quando o patrocinador do estudo de equivalência farmacêutica desenvolver metodologia local (método não oficial), ou seja, não utilizar método farmacopeico validado para o determinado produto a ser avaliado nos ensaios na equivalência, obrigatoriamente, deverá enviar ao centro de estudos toda documentação necessária. Essa documentação, são os protocolos e relatórios analíticos que são fornecidos com a finalidade de transferência da validação total ou parcial do método analítico validado pelo patrocinador, no centro de estudos (DE OLIVEIRA, et al, 2023).
A validação analítica consiste na demonstração com evidência da adequabilidade e robustez do método escolhido para o que se pretende usá-lo. São os ensaios in vitro realizados com a parametrização e equipamentos calibrados, equipe operacional qualificada desenvolvidos pelo patrocinador do estudo, testados e validados pelo centro de estudos contratado. Tais ensaios físico-químicos são fundamentais para a garantia da qualidade dos medicamentos testados. Todos os ensaios realizados no estudo de equivalência farmacêutica são documentados e ao término do estudo, toda a documentação proveniente são fornecidas à empresa patrocinadora (DE OLIVEIRA, et al, 2023).
A validação dos métodos analíticos deve ser realizada com base na resolução RDC nº 166 de 24 de julho de 2017 da ANVISA, que traz as orientações pertinentes as condições das validações a serem empregadas como parâmetros a serem avaliados, tais como: precisão, especificidade, exatidão, limite de detecção, limite de quantificação, linearidade, intervalo de aceitação e robustez (BRASIL, 2017).
3.3. BIOEQUIVALÊNCIA FARMACÊUTICA
O estudo de Bioequivalência farmacêutica também deve ser conduzido por centro de estudos habilitados que atendam boas práticas clínicas e laboratoriais. O medicamento teste e o de referência empregados no estudo de Bioequivalência devem ser os mesmos utilizados no estudo de equivalência farmacêutica (DE OLIVEIRA, et al, 2023).
O resultado do estudo é determinado pela quantificação do fármaco ou do metabólito de escolha ativo na circulação (sangue, plasma ou soro) ou quando justificado o meio escolhido, pela quantificação na urina (MENDES, 2007).
O Ensaio de Bioequivalência /Biodisponibilidade relativa é realizado em três etapas principais: etapa clínica, etapa analítica e etapa estatística, as quais devem ser conduzidas de acordo com as orientações do Guia anexo à RESOLUÇÃO-RE Nº 895, DE 29 DE MAIO DE 2003 (BRASIL, 2003).
3.3.1. ETAPA CLÍNICA
Na primeira etapa do estudo é realizado o planejamento, ou seja, o delineamento do estudo. Nessa etapa são verificados: o composto que será quantificado (analito ou metabólito), o cronograma das coletas sanguíneas conforme a forma farmacêutica (liberação imediata ou modificada), características de meia-vida de eliminação do fármaco, o número de voluntários (n), o sexo dos voluntários, a idade que varia de acordo com o objetivo do protocolo clínico, washout (período de intervalo entre a administração dos medicamentos que deve contemplar o tempo de sua eliminação), condição do estudo: se alimentado ou se jejum e a metodologia analítica para a quantificação do fármaco a coleta, preparo e armazenagem das amostras (MENDES, 2007, STORPIRTIS, 2004).
Após definidos todos os detalhes do estudo, o centro de estudos contratado deve elaborar o protocolo do estudo e submetê-lo para a aprovação do Comitê de Ética em Pesquisa (CEP) ou instituição equivalente (BRASIL, 2003. Um desenho de estudo muito aplicado é o desenho cruzado 2 x 2, proposto inicialmente por Cochram e Cox em 1962 e, desde então, é aplicado com ênfase para esses estudos (PITTA, 2004).
No protocolo do estudo deve conter todos os detalhes do estudo, como: os critérios de inclusão/exclusão, os métodos à adotar para a administração dos medicamentos aos voluntários, nesse sentido é importante ressaltar que o preconizado é que os participantes do estudo recebam a medicação ) com volume de líquido padronizado No protocolo também são inseridos os tempos de coletas das amostras para a correta avaliação dos parâmetros farmacocinéticos, dentre outros aspectos essenciais (RE nº1170, 2006[CdM8] ; MENDES, 2007).
É fundamental que o Protocolo seja aprovado pelo sistema Comitê de Ética em Pesquisa/ Comissão Nacional de ética em Pesquisa (CEP/CONEP) antes de sua condução. A Resolução 466/2012 traz várias recomendações éticas para proteção do participante de pesquisa, como a necessidade de esclarecimento de os procedimentos que serão adotados no estudo, assim como o direito de desistência a qualquer momento (RES 466, 2012).
Ainda de acordo com a RDC 742 de 2022 que dispõe sobre os critérios para a condução de estudos de biodisponibilidade relativa/bioequivalência (BD/BE) e estudos farmacocinéticos, após elaborado o protocolo do estudo, submetido e aprovado pelo CEP é necessário realizar o recrutamento dos participantes. Os procedimentos com cada participante devem ser iniciados somente após da concessão destes, com a assinatura em documento específico denominado termo de consentimento livre e esclarecido. Os participantes devem ser recrutados, identificados de acordo com os critérios de inclusão. Estes devem ser saudáveis, exceto em ensaios específicos, e a condução destes nesse processo deve ser de acordo com as regulamentações brasileiras e acordos internacionais multilaterais respeitando os valores éticos e legais (BRASIL, 2022).
É necessário que os voluntários da pesquisa sejam submetidos a avaliação clínica antes e depois do estudo de bioequivalência, abrangendo o histórico médico, exames físicos, eletrocardiograma com doze derivações e avaliações laboratoriais: sorologia, hematologia, bioquímica e urinálise. Dependendo do fármaco que será administrado podem ser necessários demais exames antes do início do estudo. Conforme o parágrafo 3º do artigo 13º da RDC 742, é recomendável os exames laboratoriais: hemograma completo; ureia; creatinina; fosfatase alcalina; glicemia; bilirrubina direta, indireta e total; proteínas totais; albumina; transaminases oxalacética e pirúvica (TGO e TGP); ácido úrico; colesterol total; triglicérides; urina tipo I (urina rotina); Beta HCG (para mulheres); sorologia para hepatite B, C e HIV (BRASIL, 2022).
Estando os participantes selecionados e clinicamente liberados para a participação no estudo, poderá ser realizado o internamento dos grupos (dois períodos) de acordo com o desenho do estudo. Após o internamento, a dieta do voluntário enquanto em estudo, dependerá da condição delineada no protocolo. Se a condição do estudo é em alimentado, o participante recebe a alimentação padronizada com base no que se confere em legislação. Se a condição do estudo for em jejum, o voluntário é mantido em jejum de no mínimo 8 horas antes do recebimento do medicamento. Importante ressaltar, que durante o estudo todos os participantes são monitorados quanto às condutas alimentares e atividade física, pois a motilidade intestinal é fator interferente para a biodisponibilidade do fármaco na circulação (PITTA, 2004).
O desenho mais usual, conforme já mencionado é desenho randomizado, cruzado 2×2, ou seja, 2 tratamentos e 2 períodos. Nesse modelo os participantes são separados em dois braços (teste e referência) e cada indivíduo se torna seu próprio controle sobre a variabilidade, permitindo a comparação entre os próprios resultados para as diferentes formulações. A administração dos medicamentos Teste e Referência, é realizada com a aleatorização apropriada (PITTA, 2004). Resume-se na figura 2, abaixo, o modelo de desenho aberto, aleatório e cruzado.
Figura 2: Desenho 2×2 para estudo de bioequivalência.
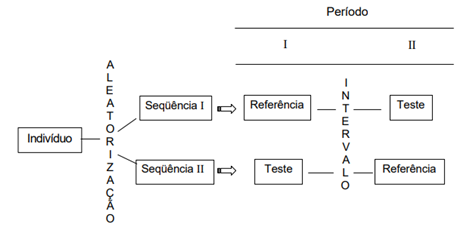
Fonte: (PITTA, 2004)
O desenho experimental que se deseja realizar deve prever uma correta definição da curva da concentração plasmática do princípio ativo versus tempo para cálculo da área sob a curva (ASC), sendo esse um dos parâmetros norteadores na determinação de biodisponibilidade e bioequivalência entre dois medicamentos (BRIOSCHI, 2007).
Durante o estudo as condições clínicas dos participantes devem ser acompanhadas e qualquer evento adverso deve ser devidamente registrado em formulário específico para registro de eventos adversos em harmonia com os procedimentos de controle ou tratamento. Caso seja necessária a introdução de medicação para o tratamento de algum efeito adverso, deverá ser avaliado a interatividade entre os fármacos na escolha do medicamento para o tratamento do evento adverso (BRASIL, 2022).
Para evitar a interferência de possíveis resíduos do medicamento do primeiro internamento da Bioequivalência, é necessário que o segundo internamento aconteça dentro de um intervalo ideal, considerando a meia-vida de eliminação. Além disso, é necessário planejar de forma adequada o armazenamento e o transporte das amostras biológicas evitando assim, interferências desse âmbito nos resultados. Orienta-se que as amostras, quando sanguíneas, sejam coletadas em tubos com anticoagulante, centrífugas e armazenadas em congelamento (BRIOSCHI, 2007).[CdM9]
Figura 03: Fluxograma da etapa clínica
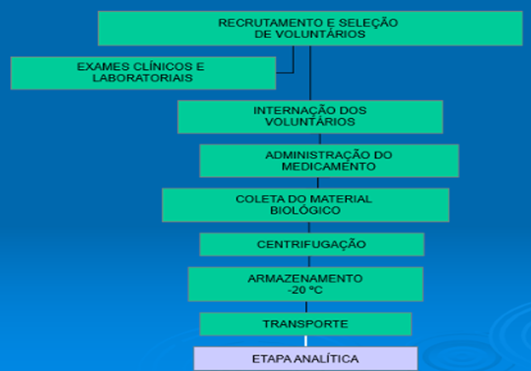
Fonte: Autores e colaboradores, 2023.
3.3.2. ETAPA ANALÍTICA
Após a coleta, preparo e armazenagem das amostras, é chegada a etapa Analítica, que por sua vez, tem o objetivo de quantificar o fármaco na matriz biológica utilizando-se de metodologia analítica validada. Para a quantificação do fármaco é indicado o uso de metodologia de simples aplicação, eficaz, segura, previamente validada e reprodutiva (BRIOSCHI, 2007).
De acordo com a Resolução 1.170 de 2006, o método bioanalítico, cromatográfico ou outro, utilizado para a quantificação da substância ativa na matriz biológica deve ser detalhadamente descrito no protocolo ou procedimento operacional Padrão (POP), validado e seguido fielmente na aplicação (RE 1.170, 2006).
Todos os procedimentos do estudo devem ser realizados de acordo com as normas internacionais de Boas Práticas de Laboratório (BPL) e conforme o GUIA PARA VALIDAÇÃO DE MÉTODOS ANALÍTICOS E BIOANALÍTICOS, e de acordo com os requisitos de biossegurança. Dentre os métodos utilizados para a quantificação do fármaco nas amostras biológicas, cita-se: Elisa, Radioimunoensaio, Cromatografia líquida de alta eficiência acoplada a espectrometria de massa s (CLAE-EM), Cromatografia líquida de alta eficiência com detecção por Ultravioleta, dentre outros. Segundo o autor, a sensibilidade e seletividade da técnica CLAE-EM possui boa reprodutibilidade em amostras de baixa concentração permitem a quantificação de amostras de baixa concentração da fase de eliminação (RE 1.170, 2006; MENDES, 2007).
Além disso, outros critérios devem ser imprescindivelmente respeitados na etapa analítica, para assegurar os resultados, tais como: estudo de estabilidade do analito (fármaco ou metabólico); O método bioanalítico deve atender a rigorosos critérios de atendimento a seletividade, efeito matriz, precisão, exatidão, estabilidade do analito em matriz biológica, sendo aplicadas curvas de calibração e controle de qualidade para garantir a qualidade da quantificação do fármaco, que se dá pela razão da área do analito pela área do padrão interno (BRIOSCHI, 2007).
Figura 04: Principais ações da Etapa Analítica.
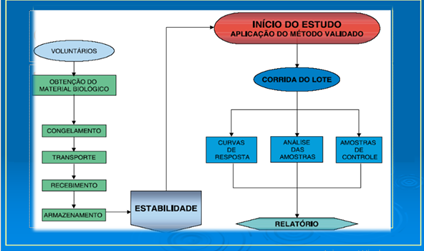
Fonte: Autores e colaboradores, 2023.
3.3.3. ETAPA ESTATÍSTICA
A etapa estatística inicia antes mesmo da etapa clínica quando se avalia o número de participantes ideal para o delineamento do estudo e tem a continuidade e a finalização após a etapa analítica (BRIOSCHI, 2007).
A análise estatística no estudo de biequivalência tem por objetivo demonstrar estatisticamente se a biodisponibilidade do medicamento teste é semelhante o suficiente ao de referência. Isso é possibilitado por meio dos parâmetros farmacocinéticos de ASC (extensão de absorção) e Cmax (velocidade de absorção). As formulações serão bioequivalentes quando o intervalo de confiança (IC) de 90% das médias geométricas da ASC e Cmax estiverem dentro do intervalo de 80-125% estabelecidos pelo FDA e ANVISA (MENDES, 2007). Este intervalo pode ser aumentado para fármacos com variabilidade maior que 30% para Cmáx, ou restringido para fármacos de janela terapêutica estreita (RDC 742/2022).
4. CLORIDRATO DE CICLOBENZAPRINA
O cloridrato de ciclobenzaprina é uma molécula sintética muito recomendada para o tratamento das dores musculares. Foi sintetizada pela primeira vez em 1960 com a finalidade de tratamento de transtornos depressivos, porém apresentou pouca ou nenhuma inovação em relação às moléculas existentes para a patologia. Com isso, passou a ser indicada para outros tratamentos, como cefaleia crônica e rigidez causada pela doença de Parkinson, ansiedade e esquizofrenia, até chegar então ser indicada para o tratamento das dores do espasmo muscular, musculoesquelética e transtornos do sono associados com a fibromialgia. Portanto, esta molécula possui alta relevância para a população, sendo importante o desenvolvimento de medicamento genérico (BRIOSCHI, 2007)
No Brasil a ciclobenzaprina é comercializada nas apresentações de 5 mg, 10 mg e 15 mg, sendo que o medicamento de referência das concentrações de 5 mg e 10 mg é o Miosan da fabricante Apsen. Já a concentração de 15 mg tem como de referência o medicamento Mitrul da fabricante Zodiac. O cloridrato de ciclobenzaprina também pode ser encontrado em associações como o Dolamin Flex 125mg + 5mg (clonixinato de lisina + cloridrato de ciclobenzaprina) da indústria de medicamentos Farmoquímica e o Miosan Caf (cloridrato de ciclobenzaprina + cafeína) nas concentrações de 5 mg + 30 mg e 10 mg + 60 mg, da fabricante Apsen (LISTA A e B – ANVISA, 2023). A dose usual recomendada diária é de 20 a 40 mg em duas a quatro administrações ao dia (a cada 12h ou a cada 6h) por via oral (BULA DIGITAL, 2023).
O cloridrato de ciclobenzaprina é um sal amino tricíclico. Apresenta-se como pó branco, livremente solúvel em soluções aquosas e alcóolicas, possui pKa de 8,47, metabolização hepática e excreção na urina. Conhecido quimicamente pela IUPAC com a nomenclatura de: 3-(5H-dibenzo, cyclohepten-5-ylidene)- N,N-dimethyl-1-propanamine. Sua estrutura molecular é apresentada na imagem abaixo (BRIOSCHI, 2007).
Figura 05: Estrutura molecular do Cloridrato de Ciclobenzaprina.
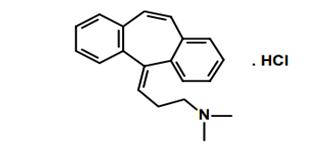
Fonte: (BRIOSCHI, 2007).
5. METODOLOGIA
O referencial bibliográfico foi realizado utilizando-se de sites e plataformas de pesquisas, tais como: Scielo, PubMed, Biblioteca Digital da USP, legislações e Resoluções de Diretoria Colegiada. Também foi utilizado de material fornecido por colaboradores ao projeto de pesquisa, mediante as autorizações prévias dos copatrocinadores.
Assim foram consultados no total 28 referências, sendo elas 7 legislações e resoluções e 28 artigos e trabalhos acadêmicos encontrados nas plataformas supracitadas. Além destes, foi consultado o material fornecido por copatrocinador da pesquisa, contendo dados reais de um estudo de Bioequivalência Farmacêutica realizado para o registro do medicamento genérico Cloridrato de Ciclobenzaprina 10 mg comprimido.
6. RESULTADOS E DISCUSSÕES: DOIS ESTUDOS DE BIOEQUIVALÊNCIA FARMACÊUTICA DO MEDICAMENTO CLORIDRATO DE CICLOBENZAPRINA 10 MG COMPRIMIDO
6.1. ETAPA CLÍNICA
Os protocolos dos estudos foram elaborados de acordo com os critérios do Guia anexado a Resolução – RE nº 896, de 29 de maio de 2003 e aprovados pelo CEP conforme Resolução 466/2012. O estudo 1 considerou em seu protocolo 36 participantes sadios, dos sexos feminino e masculino e o estudo 2 considerou 26 participantes, sendo 13 participantes do sexo masculino e 13 do sexo feminino com idade média de 32 anos. Em ambos os estudos, os voluntários participaram livre e espontaneamente, sendo orientados quanto aos procedimentos pertinentes aos critérios de exclusão.
6.1.1. INTERNAMENTO DOS GRUPOS E ADMINISTRAÇÃO DOS MEDICAMENTOS
Os voluntários foram orientados a não ingerir bebidas alcóolicas, cafés ou outras bebidas contendo xantinas, 48h antes do início dos internamentos em ambos estudos. Foram internados a noite e fizeram o jejum de no mínimo horas antes da administração dos medicamentos. As alimentações dos voluntários durante os períodos dos internamentos foram padronizadas (cada estudo teve sua dieta individual) de acordo com os critérios vigentes em legislação.
O estudo 1 foi do tipo aberto, aleatorizado, cruzado com 2 períodos de internamento, nos quais os voluntários receberam em períodos distintos a formulação teste e o medicamento de referência. As formulações foram administradas em dose única, por via oral seguidas de coletas de sangue de pelo menos 3 (três) meias-vidas. Os períodos de tratamento obedeceram a um intervalo mínimo de 7 (sete) meias-vidas entre eles (período de eliminação do fármaco pelo organismo). Considerando-se que a meia vida do Cloridrato de Ciclobenzaprina é de aproximadamente 38 (trinta e oito) horas, planejou-se um intervalo de no mínimo 15 (quinze) dias entre as internações. Os voluntários foram aleatoriamente designados a uma sequência de tratamento (T/R ou R/T T – Teste e R – Referência).
O estudo 2 foi do tipo quantitativo direto com desenho aleatório, cruzado e aberto. Os voluntários foram divididos em dois grupos A e B. No período de internamento 1, os voluntários do grupo A receberam uma dose do medicamento Flexeril, administrado como o medicamento de referência e o outro grupo recebeu o medicamento Miosan como medicamento teste. Aguardou-se 30 dias como período de washout e realizou-se o internamento do período dois em a administração dos medicamentos aos grupos foram cruzados.
6.1.2. COLETAS, ARMAZENAGEM E TRATAMENTO DAS AMOSTRAS BIOLÓGICAS
Quanto ao estudo 1, a coleta das amostras biológicas se deu início com a coleta pré-administração, realizada 60 minutos antes da administração do medicamento do experimento. Os demais horários das coletas das amostras pós-administração podem ser observados na tabela 01, abaixo. Em cada período de administração dos medicamentos teste e referência, foram coletadas 20 amostras de 7,5 mL. As amostras de sangue total foram coletadas em tubos contendo heparina sódica como anticoagulante.
Tabela 01: Horários das coletas antes e pós medicação estudo 1.
Amostra pré-administração Aproximadamente 60 minutos antes da administração da medicação Amostras pós-administração (horas: minutos) 01:00; 02:00; 02:30; 03:00; 03:30; 04:00; 04:30; 05:00; 05:30; 06:00; 06:30; 07:00; 07:30; 08:00; 09:00; 10:00; 12:00; 24:00; 48:00; 72:00.
Fonte: Copatrocinador do projeto, 2023.
No estudo 2, foram coletados 8mL de amostra biológica (sangue) dos voluntários e armazenados em tubos heparinizados. As coletas aconteceram conforme o cronograma abaixo:
Tabela 02: Horários das coletas antes e pós medicação estudo 2.
Amostras pós-administração (horas: minutos) 00:00; 01:00; 02:00; 03:00; 04:00; 05:00; 06:00; 07:00; 08:00; 10:00; 12:00; 24:00; 48:00; 72:00, 96:00, 144:00, 192:00 e 240:00.
Fonte: (BRIOSCHI, 2007) (ADAPTAÇÃO PELOS AUTORES)
Em ambos estudos as amostras foram centrifugadas e o plasma de cada voluntário foi dividido em amostra de análise e retém. As amostras de plasma foram armazenadas em temperatura adequada (-20 ºC) e encaminhadas ao laboratório analítico com os devidos cuidados quanto a estabilidade das amostras. Durante todo o período de internamento, os sinais vitais dos voluntários foram avaliados: vitais: pulso, pressão sanguínea e temperatura foram verificados aproximadamente 60 minutos antes da administração do medicamento e depois nos intervalos de administração dos medicamentos. O acompanhamento dos sinais vitais foi realizado nos dois estudos, sendo constantemente monitorados os eventos adversos.
6.2. ETAPA ANALÍTICA
Os critérios adotados pelos Centros analíticos em consideração as metodologias analíticas validadas foram definidas com base na RDC 27/2012. Os parâmetros avaliados nas ambas validações foram especificidade, precisão, exatidão, limite inferior de quantificação – LIQ, recuperação, linearidade e estabilidade. Todas as etapas foram realizadas de acordo com as Boas Práticas Laboratoriais. Abaixo nas tabelas 03 e 04 estão o resumo em relação as técnicas analíticas adotadas para os estudos 1 e 2, respectivamente.
Tabela 03: Descrição da Técnica Bioanalítica do estudo 1.
Técnica bioanalítica Cromatografia líquida acoplada à espectrometria de massas (UPLC-MS/MS) Detecção Espectrometria de massas Padrão interno Diazepam Matriz biológica Plasma Anticoagulante Heparina sódica Tipo de extração Líquido-Líquido Faixa de linearidade 0,1 – 0,5 – 1,0 – 2,5 – 5,0 – 10,0 – 20,0 – 30,0 ng/mL Limite de quantificação 0,1 ng/mL Controles de Qualidade 0,3 – 12,5 – 25,0 ng/mL
Fonte: Copatrocinador do projeto, 2023.
Tabela 04: Descrição da Técnica Bioanalítica do estudo 2.
Técnica bioanalítica Cromatografia líquida acoplada à espectrometria de massas com eletrospray-positivo Detecção Espectrometria de massas Padrão interno Amitriptilina Matriz biológica Plasma Anticoagulante Heparina sódica Tipo de extração Líquido-Líquido Faixa de linearidade 0,25 – 0,5 – 1,0 – 2,5 – 5,0 – 10,0 – 15,0 ng/mL Controles de Qualidade 0,75 – 6 – 12,0 ng/mL
Fonte: (BRIOSCHI, 2007) (ADAPTAÇÃO PELOS AUTORES)
6.3. ETAPA ESTATÍSTICA
Conforme recomendações da ANVISA e FDA os resultados obtidos nos estudos de bioequivalência foram avaliados estatisticamente de acordo com os parâmetros farmacocinéticos usuais: Cmáx, ASC0-t e ASC0-inf.
No estudo 1, foi utilizado na análise o planejamento de blocos aleatorizados modificados (crossover 2×2), no qual cada bloco recebeu mais de uma formulação de um mesmo fármaco em períodos diferentes.
Para ambos os ensaios estatísticos empregou-se a análise de variância (ANOVA) apropriada para o modelo de 2 períodos cruzados, sob os dados de ASC e Cmáx transformados logaritmicamente, a qual levou-se em conta em seu modelo os efeitos de sequência, voluntário dentro da sequência, tratamento e período.
Foram calculados os pontos paramétricos e estimativas dos intervalos da razão
para valores ASC e Cmáx. A biodisponibilidade relativa da formulação Teste versus Referência foi avaliada pelas razões das médias geométricas.
Foi construído um intervalo de confiança (IC) de 90% para a diferença das médias dos dados transformados dos medicamentos teste e referência, para os parâmetros ASC0-t, ASC0-inf e Cmáx. O antilogarítmo do IC obtido constitui o IC de 90% para a razão das médias geométricas dos parâmetros:
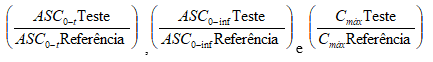
A construção desse IC foi baseada no quadrado médio residual da ANOVA. Dois medicamentos serão considerados bioequivalentes se os valores extremos do intervalo de confiança de 90% da razão das médias geométricas

forem maiores que 80% e menores que 125%.
Tabela 04: Resultados do estudo 1 de cloridrato de Ciclobenzaprina 10 mg comprimido:
Razão
T/RMédia Geométrica Intervalo de Confiança (90%) Intervalo de Bioequivalência Ln(Cmáx) 99,24 (90,70% – 108,58%) (80 % – 125%) Ln(ASC0-t) 98,68 (90,74% – 107,31%) (80 % – 125%)
Fonte: Copatrocinador do projeto, 2023.
Tabela 05: Resultados do estudo 2 de cloridrato de Ciclobenzaprina 10 mg comprimido:
Razão
T/RMédia Geométrica Intervalo de Confiança (90%) Intervalo de Bioequivalência Ln(Cmáx) 103,02 (93,00% – 112,00%) (80 % – 125%) Ln(ASC0-t) 100,69 (92,60% – 111,10%) (80 % – 125%)
Fonte: (BRIOSCHI, 2007) (ADAPTAÇÃO PELOS AUTORES)
Abaixo, apresenta-se a curva de concentração plasmática após administração oral de duas formulações de ciclobenzaprina para fins de avaliação de bioequivalência farmacêutica (BRIOSCHI, 2007).
Figura 06: Curva de concentração plasmática ao longo do tempo para formulação teste e referência de ciclobenzaprina.
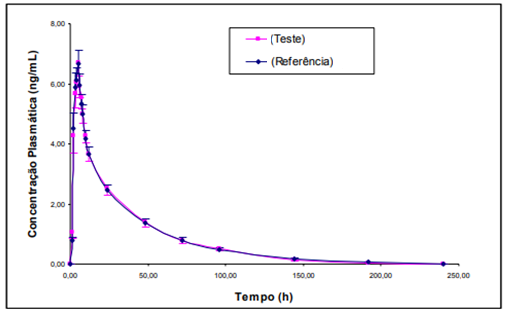
FONTE: (BRIOSCHI, 2007)
Conforme pode ser observado, ambas as curvas demonstram o comportamento farmacocinético esperado para fármacos de absorção oral, refletindo as etapas de liberação, absorção e eliminação da ciclobenzaprina, sendo semelhantes in vivo, e atendendo aos requisitos estatísticos estabelecidos em legislação.
7. CONCLUSÃO/CONSIDERAÇÕES FINAIS
Os desafios enfrentados na adoção de medicamentos genéricos, incluindo as adaptações regulatórias e a busca pela equivalência terapêutica, destacam a importância de ações que garantam a qualidade e a segurança desses medicamentos (BRASIL, 2001). A equivalência e bioequivalência farmacêutica desempenham um papel crucial na garantia da segurança e eficácia dos medicamentos, contribuindo significativamente para a promoção do acesso à saúde. Ao longo deste projeto, explorou-se os conceitos dessas duas dimensões importantes, e destacou-se a relevância na comprovação da intercambialidade entre medicamentos (ALVES et al., 2012).
Ao garantir a equivalência farmacêutica entre dois medicamentos, garante-se que diferentes formulações do mesmo princípio ativo possuam padrões de qualidade e composições semelhantes. Isso é essencial para manter a consistência no tratamento dos pacientes, evitando variações indesejadas nos resultados terapêuticos. A padronização dos produtos farmacêuticos promove a confiança tanto dos profissionais de saúde quanto dos pacientes, consolidando a eficácia do tratamento (ALVES et al., 2012).
Importante destacar que com esse projeto também foi possível verificar o essencial trabalho que é realizado pelos multiprofissionais da área da saúde, seja no desenvolvimento de novas moléculas, seja nas análises e liberações de resultados ou até mesmo in loco, na aplicação e acompanhamento dos internamentos da bioequivalência. A busca constante por métodos analíticos mais sensíveis, estudos de bioequivalência aprimorados e abordagens inovadoras são fundamentais para desenvolver alternativas eficientes para os desafios da categoria.
Vale ressaltar que é de grande valia que a comunidade científica, regulatória e industrial invista generalizadamente em pesquisa e desenvolvimento nessa área, aprimorando constantemente nossos métodos de avaliação para certificar que os pacientes tenham acesso a medicamentos seguros e eficazes.
A busca por evidências científicas sólidas nesse contexto é essencial. Estudos robustos e ensaios clínicos bem conduzidos desempenham um papel crucial na validação da equivalência e bioequivalência farmacêutica. Essa abordagem baseada em evidências não apenas fornece a segurança necessária para a intercambialidade de medicamentos, mas também contribui para a evolução contínua dos padrões de qualidade na indústria farmacêutica e para o acesso ao tratamento seguro aos pacientes (LEMES, et al, 2018).
A comprovação da intercambialidade entre medicamentos por meio da equivalência e bioequivalência farmacêutica é um pilar fundamental para garantir o acesso à saúde. A confiabilidade na prescrição, aquisição e administração dos medicamentos genéricos, fortalece a competitividade, a inovação e por conseguinte, desencadeiam melhores desempenhos da indústria farmacêutica. A oferta de medicamentos genéricos além de fornecer tratamentos eficazes, torna esses tratamentos mais acessíveis, trazendo benefícios à saúde da população consumidora (BRASIL, 2001).
Analisando-se o contexto dos detalhes empregados nas etapas de um estudo de bioequivalência é possível evidenciar que o medicamento genérico é comprovadamente um produto seguro, eficaz e que possui semelhante comportamento no organismo. Os estudos de equivalência e bioequivalência são absolutamente eficazes, pois são muito bem planejados e executados. São ensaios in vitro e in vivo muito confiáveis, regrados e proximamente acompanhados pelo maior órgão regulador do País.
Assim sendo, conclui-se que os estudos de equivalência e bioequivalência são de extrema relevância para a segurança do paciente, bem como para a ampliação do mercado econômico da classe. A compreensão abrangente dos princípios da bioequivalência e a aplicação consistente desses princípios são fundamentais para garantir que os medicamentos genéricos atendam aos mais altos padrões de qualidade.
O medicamento genérico comercializado no Brasil pode ser indubitavelmente intercambiável com o medicamento de referência, pois possui qualidade, segurança e eficácia terapêutica metodologicamente, comprovada.
8. REFERÊNCIAS
- AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA (BRASIL). Resolução-RE n° 1.170, de 19 de abril de 2006. Determina a publicação do Guia para provas de biodisponibilidade relativa/bioequivalência de medicamentos. Diário Oficial da União, 2006.
- AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA. ANVISA. Resolução RDC n. 166, de 24 de Julho de 2017. Dispõe sobre a validação de métodos analíticos e dá outras providências, 2017.
- ALVES, TEREZINHA N. P.; MATTOS, RUBEN A.; VIEIRA, R. C. P. A. Medicamentos: conceitos, usos e problemas advindos do uso. In: Convibra Saúde– Congresso Virtual Brasileiro de Educação, gestão e promoção da saúde. 2012.
- ARAÚJO, LORENA ULHÔA ET AL. Medicamentos genéricos no Brasil: panorama histórico e legislação. Revista Panamericana de Salud Pública, v. 28, n. 6, p. 480- 492, 2010.
- BERMUDEZ, J.. MEDICAMENTOS GENÉRICOS: uma alternativa para o mercado brasileiro. Cadernos de Saúde Pública, v. 10, n. 3, p. 368–378, jul. 1994.
- BRASIL. LEI Nº 6.360, DE 23 DE SETEMBRO DE 1976. Dispõe sobre a Vigilância Sanitária a que ficam sujeitos os Medicamentos, as Drogas, os Insumos Farmacêuticos e Correlatos, Cosméticos, Saneantes e Outros Produtos, e dá outras Providências. Diário Oficial da União, 1976.
- BRASIL. LEI Nº 9.787, DE 10 DE FEVEREIRO DE 1999. Lei dos medicamentos genéricos no Brasil. Poder Executivo. Brasília: MS, 1999.
- BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RE 310 de 01 de setembro de 2004. Guia para realização do estudo e elaboração do relatório de equivalência farmacêutica e perfil de dissolução. Brasília, 2004.
- BRASIL ANVISA. (2007). RESOLUÇÃO-RDC N. º 16 DE 2 DE MARÇO DE 2007.: Aprovar o Regulamento Técnico para Medicamentos Genéricos. Diário Oficial da União.
- BRASIL (2022). Ministério da Saúde/Agência Nacional de Vigilância Sanitária/Diretoria Colegiada RESOLUÇÃO – RDC Nº 749, DE 5 DE SETEMBRO DE 2022. Dispõe sobre isenção de estudos de bioequivalência /biodisponibilidade relativa.
- BRIOSCHI, Tatiane Maria de Lima Souza. Avaliação da bioequivalência de comprimidos contendo 10 mg de cloridrato de ciclobenzaprina. 2006. Dissertação (Mestrado em Produção e Controle Farmacêuticos) – Faculdade de Ciências Farmacêuticas, Universidade de São Paulo, São Paulo, 2006. doi:10.11606/D.9.2006.tde-13122006-091912. Acesso em: 2023-10-13
- CALIXTO, JOÃO B.; SIQUEIRA JUNIOR, JARBAS M. Desenvolvimento de medicamentos no Brasil: desafios. Gazeta médica da Bahia, v. 78, n. 1, 2008.
- DA SILVEIRA, NATÁLIA BRAMANO et al. Conhecimento e aceitação do medicamento genérico na população de São Miguel do Anta, MG. ANAIS SIMPAC, v. 2, n. 1, 2015.
- DE OLIVEIRA LEMES, ERICK et al. História do Medicamento Genérico no Brasil. Ensaios e Ciência C Biológicas Agrárias e da Saúde, v. 22, n. 2, p. 119-123, 2018.
- DE OLIVEIRA, Mônica Winkler et al. A equivalência farmacêutica de medicamentos genéricos e similares. Atas de Ciências da Saúde (ISSN 2448-3753), v. 11, n. 2, 2023.
- DE SOUZA VALADARES, AGNON; DO VALE, BRUNO NUNES. Medicamentos genéricos no Brasil. Revista Amazônia: Science & Health, v. 7, n. 1, 2019.
- FEDERICO, MARILIA PINTO et al. Noções sobre parâmetros farmacocinéticos/farmacodinâmicos e sua utilização na prática médica. Revista da Sociedade Brasileira de Clínica Médica, v. 15, n. 3, p. 201-205, 2017.
- MACHADO, BRUNNA GONÇALVES et al. Aceitação dos medicamentos genéricos e seus desafios: uma revisão integrativa de literatura. Research, Society and Development, v. 11, n. 8, p. e26711831133-e26711831133, 2022. file:///C:/Users/Cliente/Downloads/31133-Article-353329-1-10-20220618.pdf
- MAXIMIANO, F. P. et al.. Caracterização físico-química do fármaco antichagásico benznidazol. Química Nova, v. 33, n. 8, p. 1714–1719, 2010.
- MINISTÉRIO DA SAÚDE, BRASIL 1998. Disponível em: https://bvsms. saude. gov. br/bvs/saudelegis/gm/1998/prt3916.
- MONTEIRO, Camila Nascimento et al. Utilização de medicamentos genéricos no município de São Paulo em 2003: estudo de base populacional. Epidemiologia e Serviços de Saúde, v. 25, p. 251-258, 2016.
- NOEL, FRANÇOIS; SABINO, BRUNO DUARTE; CAMUZI, RANIERI CARVALHO. Testes de bioequivalência para fármacos que apresentem baixo índice terapêutico. Infarma, CFF, Brasília, v. 13, n. 9/10, p. 81-83, 2001.
- OLIVEIRA, E. A. DE.; LABRA, M. E.; BERMUDEZ, J.. A produção pública de medicamentos no Brasil: uma visão geral. Cadernos de Saúde Pública, v. 22, n. 11, p. 2379–2389, nov. 2006.
- OLIVEIRA, V. C. B. DE, & CAMPOS, R. (2017). Estudos de equivalência farmacêutica de comprimidos de ibuprofeno. Cadernos Da Escola De Saúde, 1(11). Recuperado de: https://portaldeperiodicos.unibrasil.com.br/index.php/cadernossaude/article/view/2402
- RUMEL, D.; NISHIOKA, S. DE A.; SANTOS, A. A. M. DOS. Intercambialidade de medicamentos: abordagem clínica e o ponto de vista do consumidor. Revista de Saúde Pública, v. 40, n. 5, p. 921–927, out. 2006.
- SILVA, ELLEN RAYNNE CALUMBY; SOUZA, THAMYRES FERNANDA MOURA PEDROSA. Aceitabilidade sobre o uso de medicamentos genéricos e seus desafios no mercado farmacêutico. Research, Society and Development, v. 11, n. 15, p. e282111537083-e282111537083, 2022.
- SIMCH, FERNANDA HELOÍSA. Testes aplicados em estudos de equivalência farmacêutica. 2013
- STORPIRTIS, SILVIA; CONSIGLIERI, VLADI OLGA. Biodisponibilidade e bioequivalência de medicamentos: aspectos fundamentais para o planejamento e execução de estudos. Rev. farm. bioquim. Univ. Säo Paulo, p. 63-70, 19
1Discente do Curso Superior de Farmácia da Universidade
União das Américas Polo Biopark – e-mail:
jaquelineterrasmendes@gmail.com
lucimarnogueira728@gmail.com
2Orientadora Profª Mestra Carin Fabíola Pensin e
Coorientadora Mestra Patrícia Moura da Rosa Zimmermann.
Docentes do Curso Superior de Graduação de Farmácia da Universidade União das Américas – Polo Biooark Educação, e-mail:
carin.pensin@bpkedu.com.br
prosfarm@hotmail.com