REGISTRO DOI: 10.69849/revistaft/fa10202501031630
Jordano Bruno Cardoso Pinto dos Santos1
RESUMO
As doenças priônicas são distúrbios neurodegenerativos raros, rapidamente progressivos e invariavelmente fatais, causados pela mutação, agregação e acúmulo da proteína priônica celular normal (PrPc), sintetizada a partir do gene PRNP no tecido nervoso. Até o presente momento, existem várias doenças causadas por príons descritas na literatura, mas a Doença de Creutzfeldt-Jakob (DCJ) segue sendo a doença priônica mais comum em seres humanos, correspondendo a mais de 85% dos casos de doenças priônicas na população mundial. A DCJ pode ser dividida em três formas: adquirida, genética e esporádica, sendo esta última o tipo mais frequente. Suas manifestações clínicas são heterogêneas, diferindo conforme a forma e o estágio da doença. O diagnóstico definitivo desta patologia é realizado através da biópsia com a identificação de príons em tecido cerebral. Conduto, em função do elevado risco de infectividade com o manuseio do material desses pacientes, é possível realizar o diagnóstico utilizando os critérios do Centers for Disease Control and Prevention (CDC), que consideram, além da sintomatologia, alterações no eletroencefalograma (EEG), ressonância magnética (RM) e marcadores no líquido cefalorraquidiano (LCR). O diagnóstico definitivo é dado pela biópsia do tecido cerebral, no entanto, devido alto risco de contaminação e morbidade para tal procedimento, lançamos mão dos critérios clínicos, radiológicos e eletroencefalográficos para corroborar a hipótese diagnóstica.
O tratamento atual da DCJ é apenas de suporte e seu desfecho é fatal, sendo que a maioria dos doentes evolui para óbito em até um anodo início dos sintomas. O suporte familiar, juntamente com um acompanhamento multidisciplinar é essencial para diminuir o sofrimento e melhorar a qualidade de vida do paciente. O presente estudo, buscou relatar o caso de uma paciente de 72 anos, sexo feminino e sem comorbidades prévias relatas, moradora da capital São Luís (MA). Diagnosticada com DCJ com a forma esporádica, sinais e sintomas, assim como métodos diagnósticos e tratamento através de revisão de literatura.
Palavras-chave: Doenças priônicas; Proteínas priônicas; Síndrome de Creutzfeldt-Jakob; Demência; Encefalopatias; Relatos de casos.
ABSTRACT
Prion diseases are rare, rapidly progressive neurodegenerative disorders and Invariably fatal, caused by the mutation, aggregation and accumulation of prion protein Normal cell (PrPc), synthesized from the PRNP gene in nervous tissue. Until the present. At the moment, there are several diseases caused by prions described in the literature, but Creutzfeldt-Jakob Disease (DCJ) is still the most common prion disease in humans, Corresponding to more than 85% of the cases of prionic diseases in the world’s population. The DCJ can be divided into three forms: acquired, genetic and sporadic, the latter being the type More frequent. Its clinical manifestations are heterogeneous, differing according to the form and the stage of the disease. The definitive diagnosis of this pathology is made through biopsy with The identification of prions in brain tissue. Conduit, due to the high risk of infectivity with the handling of the material of these patients, it is possible to make the diagnosis using the criteria of the Centers for Disease Control and Prevention (CDC), which consider, in addition to symptomatology, changes in the electroencephalogram (EEG), magnetic resonance imaging (MRI) and markers in the cerebrospinal fluid (CSF). The definitive diagnosis is given by the biopsy of the brain tissue, however, due to the high risk of contamination and morbidity for such a procedure, we use the clinical, radiological and criteria to corroborate the diagnostic hypothesis.
The current treatment of CJD is only unsupporting and its outcome is fatal, and most patients progress to death within one year of the onset of symptoms. Family support, together with a multidisciplinary follow-up is essential to reduce suffering and improve the patient’s quality of life. The present study sought to report the case of a 72-year-old patient, female and without previous comorbidities reported, resident of the capital São Luís (MA). Diagnosed with JCD with sporadic form, signs and symptoms, as well as diagnostic methods and treatment through literature review.
INTRODUÇÃO
As doenças priônicas são distúrbios neurodegenerativos raros, rapidamenteprogressivos e invariavelmente fatais, causados pela mutação, agregação e acúmulo da proteína priônica celular normal (PrPc), sintetizada a partir do gene PRNP no tecido nervoso (Haddad NM, 2022). Tanto a PrPc, como a sua isoforma patogênica (PrPSc), possuem a mesma estrutura primária, mas suas estruturas secundárias diferem, com um predomínio de cadeias beta na forma patogênica. Estas alterações estruturais conduzem a perda neuronal, astrocitose e alterações espongiformes no sistema nervoso central (SNC), razão pela qual as doenças priônicas também podem ser denominadas de encefalopatias espongiformes. (Penna GL, 2017).
A DCJ, ou encefalopatia espongiforme, foi descrita pela primeira vez em 1920 por H.G. Creutzfeldt e A. Jakob. (Henry R, 2017). Historicamente, propunha-se uma etiologia viral de evolução lenta para a doença, porém os achados histopatológicos não se relacionavam a tal etiologia. Somente em 1960, Stanley Prusiner, pela primeira vez, definiu o termo príon e o atribuiu como agente envolvido na fisiopatologia da DCJ.(Cardoso CA, 2015).
As manifestações clínicas da DCJ são heterogêneas e diferem conforme o estágio da doença. Na fase inicial, os sintomas são inespecíficos e costumam ser relatados como fadiga, vertigem ou alterações de comportamento. Com o passar das semanas, o sintoma mais prevalente é a deterioração das funções cognitivas de maneira rápida e progressiva, com o desenvolvimento de demência em poucas semanas. Sintomas cerebelares também podem ser encontrados nesta fase, usualmente associados à queixa de desequilíbrio, caracterizados como ataxia da marcha.(Mendes Pires M, 2020). Conforme a doença progride, 90% dos pacientes apresentam mioclonias, sintomas piramidais e extrapiramidais, evoluindo para um estado de mutismo acinético em poucos meses.(Fagundes De Oliveira M, 2018).
O modo de transmissão natural da CJD também permanece um mistério. Vários mecanismos hipotéticos para a origem e propagação da CJD têm sido propostos, incluindo a exposição à carne de gado infectada (consistindo na variante da CJD), transmissão iatrogênica (minoria atualmente), mutações somáticas espontâneas (constituindo a CJD familiar em “clusters”) (Prusiner SB, 1993). A forma familiar da CJD é herdada com padrão autossômico dominante com mutações definidas no gene produtor da proteína (PrPC).(Tyler KL, Martin JB,1993.) Mas a maioria dos casos de CJD são esporádicos e não mostram relação com mutações específicas. Nestes casos, observou-se que a homozigose para o polimorfismo no códon 129 do gene produtor da proteína PrPC pode aumentar a suscetibilidade à infecção pelo príon (acredita-se que esta homozigose pode aumentar a conversão do PrPC para PrPSC).
A fisiopatologia da DCJ se baseia na conversão do príon do hospedeiro (PrP) em uma isoforma patogênica (PrPSc) por um desdobramento anormal. (Penna GL, 2017). A isoforma PrPsc está associada a depósito de placas amiloides nas regiões sinápticas e perivasculares, causando lesões espongiformes no encéfalo, preferencialmente nos gânglios da base, tálamo, cerebelo e córtex cerebral.(Fraser PE, 2014).
Relato de Caso Clínico
Paciente do sexo feminino, 72 anos, natural e residente em São Luís (MA), com diagnóstico prévio de hipertensão arterial sistêmica (HAS) e diabetes mellitus tipo 2 (DM2).
A paciente foi admitida na emergência do Hospital UDI (São Luis, MA), com queixa de cefaléia há 10 dias associada à perda de força motora em hemicorpo direito e disartria. Referia início súbito dos sintomas de incoordenação motora e alterações na fala há 4 dias. Sem queixas de febre, crises convulsivas ou outros sintomas associados. Foi então realizada tomografia de crânio, com hipótese diagnóstica de acidente vascular encefálico (AVE), no entanto, com resultado negativo à priori.
Em sua avaliação com a equipe de neurologia, aventa-se possibilidade de acidente vascular isquêmico de fossa posterior, sendo realizado Ressonância magnética (RM) de crânio, mas sem confirmação da hipótese e com achados típicos a seguir;
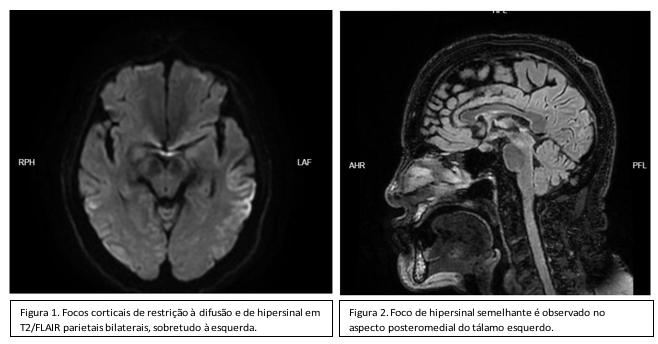
O quadro foi interpretado como demência rapidamente progressiva, e a hipótese de DCJ foi aventada. O liquor céfalo raquidiano (LCR) foi submetido a testes de reação em cadeia da polimerase (PCR) para varicela-zóster, Epstein-Barr, herpes simples 1 e 2, citomegalovírus e tuberculose, os quais foram negativos. O eletroencefalograma (EEG) não registrou nenhuma alteração compatível com o quadro clínico em questão. A função tireoidiana e a vitamina B12 estavam normais, e Veneral Disease Research Laboratory (VDRL) e anti-HIV foram não reagentes. Fora solicitado então, proteína 14.3.3, que evidenciou resultado de 31.633 (VR < 20.000). Após 4 semanas de evolução, já sob cuidados paliativos exclusivos. Evoluiu com mutismo acinético e degradação do estado geral, culminando com óbito.
DISCUSSÃO
De acordo com a Organização Mundial da Saúde (OMS), a incidência anual de DCJ é de cerca de 1,5 milhão em todo mundo, o que a caracteriza como rara e, consequentemente, de baixo índice de suspeição clínica. Esse fato, aliado à falta de conhecimento sobre a doença, torna seu diagnóstico um desafio.Tal raridade pode ser observada no estudo de Cardoso et al (Cardoso CA, 2015) que, por meio de uma avaliação epidemiológica, revelou que foram identificados apenas 132 casos de DCJ no Brasil durante o período de janeiro de 2005 a dezembro de 2010.(Cardoso CA, 2015).
A DCJ pode apresentar-se de três formas: a forma esporádica, a familiar e a iatrogênica. A forma esporádica da DCJ (DCJe), é a responsável por 85% dos casos e não está relacionada a nenhuma evidência de transmissibilidade, devido à baixa infectividade do agente. A forma familiar (DCJf), equivale a 10% dos casos e apresenta padrão autossômico dominante de herança. Já a forma iatrogênica (DCJi) atualmente corresponde a menos de 1% dos casos e é a variável que possui seu mecanismo mais bem estabelecido pela literatura. A DCJi é causada por inoculação de príons em materiais contaminados, podendo ser instrumentos cirúrgicos, enxertos de dura-máter, transplante de córnea ou transfusões de sangue. (Muniz BC, 2019). A doença, que tem característica transmissível aos humanos, é a chamada de nova variante da DCJ e está associada ao consumo de carne ou subprodutos de bovinos contaminados (doença da vaca louca).(Cardoso CA, 2015).
A apresentação clínica inicial da DCJ é inespecífica, com aspecto central de uma demência progressiva rápida, que cursa com achados neurológicos multifocais, como ataxia, afasia, perda visual, hemiparesia, amiotrofia e mioclonias.(Penna GL, 2017, Venneti, 2010, Kojima G, 2013). Além desses, alguns sintomas prodrômicos, como astenia, cefaleia, alucinações e alterações do comportamento do sono e do apetite, podem ser notados. A doença progride com demência na maioria dos casos.(González MG, 2015, Neitzke I, 2009).
O diagnóstico definitivo da doença, é dado por meio de identificação histopatologia através de biópsia ou autópsia do cérebro, obtido após a morte. Onde é possível observar disposição patológica de proteínas priônicas no tecido cerebral, caracterizada por alterações espongiformes, perda neuronal e glicose. É obrigatório a identificação da PrPsc para confirmação do diagnóstico (González MG, 2015, Penna GL, 2017). Existem critérios diagnósticos definidos pela OMS desde 1998, adaptadas pelo Centers for Disease Control and Prevention (Kojima G, 2013). Tais critérios dividem os casos suspeitos em diagnósticos definitivo, provável e possível, de acordo com a associação dos acha dos clínicos e de exames complementares (Tabela 1).
Exames complementares, como RNM, EEG e coleta de LCR são úteis no auxílio diagnóstico, apesar de não serem específicos para a doença. No entanto, podem ajudar em diagnósticos diferencias durante o período de elucidação. Assim como, mensurar complicações inerentes ao processo natural da patologia. (Venneti, 2010, Kojima G, 2013). Na RM, alterações de sinal em núcleos caudado e/ou putâmen, na sequência de recuperação de inversão atenuada de fluido (FLAIR), e sinais de hiperintensidade em lóbulos parietais e temporais bilaterais e tálamo, nas sequências ponderada em DWI e T2, são achados típicos e frequentemente observados em pacientes com DCJ (González MG, 2015). Muito raramente a análise do LCR apresenta alguma anormalidade significativa, mas é útil para descartar encefalites e outras neuroinfecções. (Venneti, 2010, Cardoso CA, 2015). O EEG pode evidenciar complexos periódicos de ondas agudas, os quais são encontrados em até 66% dos pacientes com DCJ. (Neitzke I,2009, Kojima G, 2013). Quando presente no contexto clínico da doença, o teste de Western blot para pesquisa de fragmentos de proteína 14-3-3 no LCR pode chegar a 96% de sensibilidade e 99% de especificidade (Penna GL, 2017, Kojima G, 2013). Sendo assim, o diagnóstico é realizado por meio de achados clínicos compatíveis com a doença, além de alterações na RNM, EEG e LCR.
O curso da DCJ segue rápida evolução e piora do status cognitivo e funcional, em direção ao mutismo acnético em estágio avançado. E invariavelmente para a morte em 100% dos casos. Atualmente não conhecemos tratamentos específicos para a doença, sendo o diagnóstico precoce uma chave fundamental para controle dos sintomas.
Tabela 1. Critérios diagnósticos para elucidação dos casos suspeitos de forma esporádica de doença de Creutzfeldt-Jakob
Definitivo: detecção de proteína priônica protease resistente e/ou presença de fibrila associada aos scrapies por meio de neuropatologia, técnica imunoquímica e/ou Western blot. Provável: nenhum achado indicando diagnósticos alternativos + demência progressiva com pelo menos 2 de I a IV e pelo menos um de A C. Possível: nenhum achado que indique diagnósticos alternativos + demência progressiva com duração inferior a 2 anos + pelo menos 2 de I a IV e pelo menos um de A C. (I) Mioclonias
(II) Problemas visuais ou cerebelares
(III) Sinais piramidais ou extrapiramidais
(IV) Mutismo acinético
(A) Complexos periódicos de ondas agudas no eletroencefalograma
(B) Proteína 14-3-3 positiva no líquido cefalorraquidiano com duração de doença inferior a 2 anos
(C) Anormalidades de hipersinal no núcleo caudado e/ou putâmen na imagem ponderada por difusão (DWI) ou recuperação de inversão atenuada por fluido (FLAIR) na ressonância magnética.
No referido relato de caso, optou-se por não ser realizado biópsia para confirmação diagnóstica, levando em consideração os sinais e sintomas apresentados, além da dosagem dos níveis de proteína 14.3.3 e exames de imagem complementares. Sendo considerados suficientes para elucidação do caso e diagnósticos diferenciais.
A DCJ é uma encefalopatia espongiforme rara, neurodegenerativa e de manifestação clínica múltipla, mas que deve ser sempre colocada como diagnóstico diferencial em quadros de demência de rápida progressão. Os exames complementares são essenciais para identificação dos casos suspeitos, além de possibilitarem a exclusão de outras doenças. Quando o doente não tem história pessoal de exposição, como iatrogenia, e nem consumo de carne, ou subprodutos de bovinos contaminados, o quadro pode favorecer o diagnóstico de e DCJ.
REFERÊNCIAS
- Haddad NM, Oliveira HP, Marzani LE, Frainer DA, Welter C da S. DOENÇA DE CREUTZFELDT-JAKOB INICIANDO COMO UM SURTO PSICÓTICO: UM RELATO DE CASO. Brasília Médica. 2022;59.
- Penna GL de A, Filho RC de O, Augusto M, Kaliszchtein M, Nobre G. Doença de Creutzfeldt-Jakob: forma esporádica: relato de caso em paciente de 81 anos. Revista da Sociedade Brasileira de Clínica Médica [Internet]. 2017 Nov 9 [cited 2022 Dec 31];15(3):188– 91. Available from: https://www.sbcm.org.br/ojs3/index.php/rsbcm/article/view/294
- Rui M, Salvador A. UNIVERSIDADE DA BEIRA INTERIOR Ciências da Saúde Doença de Creutzfeldt-Jakob A propósito de um caso clínico [Internet]. Available from: https://ubibliorum.ubi.pt/bitstream/10400.6/4991/1/3264_6626.pdf
- Henry R, Murphy FA. Etymologia: Creutzfeldt-Jakob Disease. Emerg Infect Dis. 2017;23(6):956.
- Cardoso CA, Navarro MB, Soares BE, Cardoso TA. Avaliação epidemiológica dos óbitos por doenças priônicas no Brasil sob o enfoque da biossegurança. Cad Saúde Colet. 2015;23(1):2-10.
- Mendes Pires M, Naiara Costa Ribeiro I, Santana Santos Queiroz T, Seixas Fukuda J, Fukuda T, Pereira de Jesus PA. Doença de Creutzfeldt-Jakob, Variante de Heidenhain. Relato de Caso e Revisão do Tema. Revista Científica Hospital Santa Izabel. 2020 May 16;1(1):30–
- Fagundes De Oliveira M, Devens V, Luis T, Anflor C, Rubião J, Filho H. ASPECTOS CLÍNICOS E RADIOLÓGICOS DA DOENÇA DE CREUTZFELDT- JAKOB [Internet]. Available from: https://docs.bvsalud.org/biblioref/2018/03/880051/aspectos-clinicos-e- radiologicos-da-doenca-de-creutzfeldt-jakob.pdf.
- Prusiner SB. Genetic and infectious prion diseases. Arch Neurol 1993; 50:177-188.
- Tyler KL, Martin JB. Infections diseases of the central nervous system. Philadelphia: F.A. Davis 1993.
- Fraser PE. Prions and prion-like proteins. J Biol Chem. 2014; 289(29):19839-40.
- Muniz BC, Makita LS, Ribeiro BN de F, Marchiori E. The Heidenhain variant ofCreutzfeldtJakob disease. Radiologia Brasileira. 2019 Jun;52(3):199–200.
- Venneti S. Prion diseases. Clin Lab Med. 2010;30(1):293-309.
- Kojima G, Tatsuno BK, Inaba M, Velligas S, Masaki K, Liow KK. Creutzfeldt-Jakob Disease: a case report and differential diagnoses. Hawaii J Med Public Health. 2013;72(4):136-9.
- González MG, Marín AG, Vargas NP, Beltrán CE, Amaya EV. Forma esporádica de enfermedad de Creutzfeldt-Jakob: reporte de dos casos. Acta Neurol Colomb. 2015;31(3):291-8.
- Neitzke I, Brito HF, Brandão A, Narciso-Schiavon JL, Schiavon LL, Buzzoleti FC. Apresentação clínica da Doença de Creutzfeldt- Jakob como síndrome cerebelar. Rev Neurocienc. 2009;17(1):63-6.
1Graduado em Medicina – Centro Universitário do Pará – CESUPA
Especializando em Medicina Intensiva – UDI Hospital Rede D’or (2022-2025)
Título de especialista em Medicina Intensiva – AMIB (2024)
Pós Graduado em Medicina Intensiva – ENSINE – AMIB (2022)
Pós Graduando em Neurointensivismo – CCD