CREUTZFELDT-JAKOB DISEASE IN BRAZIL: A REVIEW OF ITS PROFILE AND NOTIFICATION MECHANISMS.
REGISTRO DOI: 10.5281/zenodo.12086373
Vinícius Bacelar Ferreira1
Érica Giovanna de Souza Biváqua2
Felipe Alves de Paiva3
Pietra Amorim Cerquinho Oliveira4
Riesly de Oliveira Vasconcelos5
Ronilson Ferreira Freitas6
RESUMO
A Doença de Creutzfeldt-Jakob (CJD) é uma condição neurodegenerativa de natureza rara e inevitavelmente letal, caracterizada por uma progressão rápida de demência. A CJD está na lista de doenças de notificação compulsória do Ministério da Saúde no Brasil e embora se apresente como um potencial risco econômico e de saúde, há falta de dados específicos sobre a doença no Brasil. Este estudo objetivou analisar o perfil da CJD no Brasil com o objetivo de ampliar a compreensão do padrão epidemiológico da doença no país e suas características distintas, utilizando os motores de busca PubMED e SciELO a fim de buscar artigos relacionados ao tema, enquanto a incidência da doença no Brasil foi baseada em dados acessíveis através do Sistema de Informação de Mortalidade (SIM) e do DATASUS, com registros das dificuldades de acesso. Inicialmente 35 artigos foram selecionados para análise, dos quais 15 preencheram os critérios estabelecidos com 18 casos totais. Os resultados revelam que a CJD afeta predominantemente pessoas brancas do sexo feminino, com maior incidência na faixa etária de 60 a 69 anos A região Sudeste do Brasil é a mais afetada, principalmente nos estados de São Paulo, Rio de Janeiro e Minas Gerais. Além disso, foi possível estabelecer que o caráter da doença como notificação compulsória tem sido benéfico para o aumento da notificação, porém há algumas divergências do protocolo nacional se comparado com o padrão internacional que devem ser consideradas.
Palavras-chave: Príons, Doenças Priônicas, Doença de Creutzfeldt-Jacob.
ABSTRACT
Creutzfeldt-Jakob Disease (CJD) is a rare and fatal neurodegenerative condition, characterized by a rapidly progressive dementia. CJD is on a compulsory notification list of diseases by the Health Ministry in Brazil and, even though it appears as a potential economic and health risk, it lacks specific data about the disease in Brazil. This paper aims to analyze the CJD profile in Brazil, with the objective to broaden the knowledge of its epidemiological pattern in the country and its distinct characteristics, utilizing as search engine PubMED e SciELO to search for papers related to the theme, while the incidence of the disease in Brazil was based in data accessible via the Sistema de Informação de Mortalidade (SIM) and from DATASUS, with some difficulties to access. Initially 35 articles were selected to analyze, from which only 15 filled out the criteria established with 18 cases total. The results revealed that CJD affects predominantly white people, females with greater incidence in the age range of 60 to 69 years. The southeast region of Brazil is the most affected, principally the states of São Paulo, Rio de Janeiro and Minas Gerais. Furthermore, it was possible to establish that featuring the disease as compulsory notification has been beneficial to raise notification, although there are some divergences in the national protocol, if compared with the international standard, that should be considered.
Keywords: Príons, Prionic Diseases, Creutzfeldt-Jakob Disease.
1. INTRODUÇÃO
A Doença de Creutzfeldt-Jakob (CJD) é caracterizada por demência rapidamente progressiva e inevitavelmente fatal. Sendo causada por uma proteína mal enovelada conhecida como Príon, que infecta regiões altas do encéfalo a posteriores ao olho, resultando em sintomas neurológicos e alterações oculares (1).
Apesar de serem patologias de raridade acentuada, doenças priônicas são passíveis de transmissão, de modo com que epidemias que afetam tanto humanos como animais emergiram globalmente nas últimas cinco décadas, importante ressaltar que dentre as diversas doenças priônicas a de maior relevância é a CJD (2).
A CJD possui três formas: Esporádica, Familiar e Adquirida. A forma mais comum é a esporádica, correspondendo a aproximadamente 85% dos casos de CJD e afeta principalmente homens a partir dos 67 anos, a transformação da proteína PrP que ocorre nessa forma é por mecanismos ainda obscuros. Por outro lado, a forma familiar ou genética já foi demonstrada possuir relação com mutações no gene PRNP em especial do códon 129 (3), essa forma corresponde a, aproximadamente, 10-15% do total de casos (1).
Por fim, a forma adquirida, caracterizada pela infecção do indivíduo pelo agente priônico, conta com alguns subtipos, dentre os quais se incluem a Kuru, forma relacionada com rituais canibais de regiões do encéfalo; a forma Iatrogênica, devido ao agente priônico ser de difícil eliminação dos equipamentos neurocirúrgicos, apresenta riscos de ser transmitida a partir de neurocirurgias e há a forma Variante caracterizada pelo consumo de carne bovina contaminada. A forma adquirida é a forma mais rara da doença em humanos (1).
Diante de tal perspectiva e seu caráter fatal, a CJD integra a Lista das Doenças e Notificação compulsória do Ministério da Saúde, definida pela Portaria de Consolidação GM/MS nº 4, de 28 de setembro de 2017 (4). Ainda assim, apesar de compor tal lista, carecem de dados específicos sobre a doença em território brasileiro, de modo que o Brasil é notavelmente deixado de fora de estudos internacionais relevantes (1,3). Tal exclusão é preocupante, em especial, devido ao caráter agroexportador do Brasil, haja visto que a Doença da Vaca Louca corresponde a um subtipo de Doença de Creutzfeldt-Jakob, podendo ocorrer o contágio pela carne bovina contaminada e isso até mesmo impacta nas exportações do país (5,6).
A incidência global de CJD é crescente, em especial da forma esporádica, podendo indicar riscos para os sistemas públicos no futuro, de modo que estudos e sistemas de vigilâncias são uma necessidade para a avaliação longitudinal e cuidadosa da doença (3).
Logo, diante do crescimento global de casos e o potencial zoonótico da DCJ, a abstinência de informações sobre essa doença no Brasil é preocupante, haja visto os danos econômicos e salutares que podem ser ocasionados pela mesma, demonstrando que um controle de casos é uma necessidade reconhecida pelo Ministério da Saúde. Esse artigo então possui o intuito de traçar um perfil da CJD no Brasil, através dos relatos de casos encontrados por motores de buscas, assim como as informações contidas nos bancos de dados do Sistema Único de Saúde (SUS), para então, avaliar os mecanismos e protocolos de notificação no país.
2. OBJETIVO
Estabelecer as principais características da população afetada pela Doença de Creutzfeldt-Jakob no Brasil e avaliar seus mecanismos de notificação.
3. METODOLOGIA
3.1 CARACTERIZAÇÃO DO ESTUDO
Trata-se de um estudo descritivo, exploratório com abordagem qualitativa e quantitativa, cuja pergunta de pesquisa foi: “Quais são as diferenças e semelhanças do perfil da Doença de Creutzfeldt-Jakob e de seus mecanismos de notificação que acontecem no Brasil se comparado internacionalmente?”. O acrônimo PECO foi utilizado como base para a montar essa pergunta: P (População)- pacientes portadores de CJD; E (Exposição)- Naturais e procedentes do Brasil; C (Comparação)- Dados das características mundiais dos portadores de CJD; O (Outcome/Resultado): Semelhanças e diferenças dos portadores de CJD brasileiros com os internacionais.
O estudo valeu-se de relatos de casos disponibilizados em bases de dados, assim como de dados secundários disponibilizados provenientes do Sistema de Informações sobre Mortalidade (SIM) do Departamento de Informática do Sistema Único de Saúde (DATASUS), do Ministério de saúde (MS), sobre a mortalidade do CID A81 que compreende a CJD e outras doenças raras como a Leucoencefalopatia multifocal progressiva e a Panencefalite esclerosante subaguda.
3.2 LEVANTAMENTO DE DADOS BIBLIOGRÁFICOS
Para a avaliação dos relatos de casos no Brasil, a técnica da Revisão Integrativa foi utilizada. Para o levantamento bibliográfico se utilizou os seguintes motores de busca: PubMED (que fornece acesso ao banco de dados MEDLINE) e o Scientific Electronic Library Online (SciELO). A escolha dessas bases se deu pelo fato de estas fornecerem ampla literatura em diferentes áreas da saúde, possibilitando um acesso maior aos diversos artigos científicos publicados sobre esta síndrome rara. A escolha dos artigos seguiu as diretrizes do Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA). Sendo, portanto, realizado uma busca em 4 etapas: Identificação (Busca inicial contemplando todas os artigos encontrados através das palavras de busca), Triagem (Remoção de artigos incompatíveis com a pergunta de pesquisa por título ou resumo), Elegibilidade (Etapa na qual ocorrerá a aplicação dos critérios de inclusão ou exclusão) e a Inclusão (Etapa na qual estabeleceu-se o corpus do trabalho).
Os artigos foram buscados durante o período de julho de 2023 até agosto de 2023, utilizando os seguintes descritores “Creutzfeldt-Jakob Disease”; “Case Reports” e “Brazil”, unidos pelo conectivo “AND”. Após a inserção desses termos e realizada foi possível realizar a identificação dos artigos. Após essa identificação, foram removidos os artigos duplicados e realizada uma Triagem por título e resumo, removendo os artigos que já indicavam relatos de doenças mimetizando a CJD, ou com apresentação semelhante, ou que não abordassem a CJD. Após a triagem, realizou-se a avaliação da Elegibilidade dos artigos através de critérios de inclusão e exclusão.
Os critérios de inclusão foram: (1) os estudos devem estar escritos em português, inglês ou espanhol, (2) devem abordar um caso confirmado de Doença de Creutzfeldt-Jakob, (3) devem descrever tais casos com ocorrência em território brasileiro. Os critérios de exclusão foram: (1) artigos que não possuíam resumo disponível na base de dados serão excluídos, (2) abordassem outras doenças com apresentação semelhante a Doença de Creutzfeldt-Jakob; (3) não fossem relatos de casos serão desconsiderados (4) não puderam ser acessados pelo Sistema Integrado de Bibliotecas da Universidade Federal do Amazonas (SISBIB/Ufam).
Utilizando tais critérios, os trabalhos foram selecionados, dessa maneira foi estabelecido o corpus do trabalho na etapa da Inclusão. Após estabelecer o corpus do trabalho, os artigos foram lidos e analisados, e assim identificados as principais características dos pacientes acometidos pela doença, podendo avaliar-se um perfil de risco.
Ademais realizou-se uma revisão de literatura das diretrizes brasileira acerca da notificação da doença, encontradas no “Protocolo de notificação e investigação da doença de Creutzfeldt-Jakob com foco na identificação da nova variante” protocolado pelo Ministério da Saúde Brasileiro em 2018, disponibilizado de forma gratuita e virtual no endereço eletrônico: https://www.gov.br/saude/pt-br/centrais-de-conteudo/publicacoes/s vsa/doenca-de-creutzfeldt-jakob/protocolo_notificacao_investigacao_doenca_creutzfeld t_jakob.pdf/view. Comparando-a com as propostas internacionais preconizadas encontradas nos artigos de Uttley et al(1) e Watson et al(3), dessa maneira avaliando a efetividade das medidas tomadas no Brasil se comparadas ao resto do mundo, quanto às medidas e protocolos base para a notificação da Doença de Creutzfeldt-Jakob.
3.3 LEVANTAMENTO DE DADOS SECUNDÁRIOS
Para a avaliação do número total de casos notificados da doença no Brasil, se baseia nos dados possíveis de serem acessados pelo sistema TabNET assim como outros sistemas disponíveis pelo DATASUS, em especial as informações foram colhidas através do Sistema de Informação de Mortalidade (SIM); durante tal busca, as dificuldades de acesso e outros empecilhos foram registrados, para a avaliação do acesso à informação acerca da doença. Essas informações foram colhidas através do CID A81 que compreende a Doença de Creutzfeldt-Jakob e outras 4 categorias de doenças, portanto as análises retiradas do DATASUS servem como uma aproximação da situação real. A escolha da mortalidade permite uma estimativa melhor da dimensão da CJD, tendo em vista seu caráter inevitavelmente fatal, sendo por isso selecionada como parâmetro principal desse trabalho. Desse modo, na busca foi selecionado a categoria CID-10 como elemento compositor da linha da pesquisa, especificando para o CID A81 e para os elementos da coluna tomou-se como referência as seguintes variáveis: a faixa etária, o sexo, a raça e a região geográfica, sendo por fim o conteúdo selecionado foi óbitos por residência. Utilizando esses parâmetros foram avaliados três períodos: inicialmente de 1996-2017, anterior a diretriz de notificação compulsória da CJD estipulada pelo Ministério da Saúde e o período de 2018-2021, após a obrigatoriedade de notificação compulsória e o período geral compreendido de 1996-2021.
4. RESULTADOS
A figura 1 mostra o processo de seleção dos artigos, constando o padrão da busca e a exclusão de artigos com suas razões específicas. Inicialmente foi encontrado um total de 35 artigos em ambas as bases de dados, dos quais 8 eram duplicados, isto é, estavam presentes em ambas as plataformas, de modo que o total sem duplicatas é 27. Após a leitura preliminar por título e resumo eliminou-se 9 artigos, devido a apontarem que não eram relato de caso ou que abordavam doenças que não fosse CJD, sendo notável por título e resumo, assim foram triados um total de 18 artigos, avaliados na íntegra e restando ao final do processo 15 relatos de caso que foram incluídos nesta análise; sendo que os 3 artigos que foram excluídos, eram por ser: um artigo apresentar um caso não-brasileiro e outros dois apresentarem doenças símiles a CJD.
Figura 1: Processo de seleção de artigos.
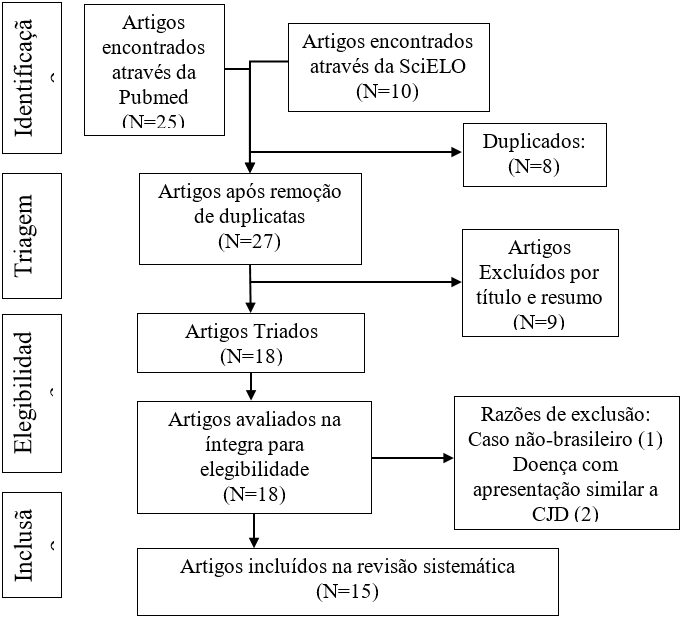
Fonte: Elaborado pelos autores, 2023.
Após a construção do corpus de relatos de casos, extraiu-se dados relevantes para a análise apresentados na Tabela 1.
Ao total foram contabilizados 18 casos presentes nos 15 artigos, sendo possível identificar o sexo dos pacientes, a idade de uma maneira geral, em alguns foi possível identificar a forma da CJD, alguns dos hábitos de vida, biomarcadores, naturalidade e etnia, sendo que os achados mais relevantes identificados foram o sexo e a idade, colunas 2 e 3 da Tabela 1, respectivamente.
A média das idades foi aproximadamente 54 anos, sem quaisquer diferenças significativas entre os sexos. A doença foi mais frequente, no sexo masculino que foi responsável por 77,7% dos casos analisados em sua maioria da etnia branca e sendo encontrado mutação genética em 38,8% dos casos (Tabela 1).
Tabela 1: Características dos pacientes portadores de CJD encontrada nos relatos de casos.
Autor Sexo Idade Forma relatada Hábitos de Vida Biomarcador Naturalidade Etnia Ano de publicação Arruda et.al (7) Mulher 54 Não relatado Obesa Polimorfismo do códon 129 do gene PRPN. Ponta grossa, PR Branca 2004 Azevedo et.al (8) Mulher 42 Não relatado Profissão: do lar Não relatado Belém, Pará Branca 2001 Brito-Marques et.al (9) Homem 61 Não relatado Não relatado Não encontrado Não informado Não informado 2021 Ciarlariello et.al (10) Homem 56 Não relatado Não relatado Não encontrado Não informado Branca 2018 Dahy et.al(11) Homem 52 Possivelmente esporádica Não relatado Não encontrado Não informado Não informado 2021 Dahy et.al (11) Homem 61 Não relatado Não relatado Não relatado Não informado Não informado 2021 De souza Et.al (12) Mulher 57 Não relatado Não relatado gCJD V180I mutation Não informado Branca 2017 Gomes et al (13) Homem 52 Esporádica Não relatado Não encontrado Não informado Branca 2019 Huang et.al (14) Mulher 57 Familiar Não relatado V210I mutation Alemanha, residente no Brasil Branca 2001 Nitrini et.al (15) Mulher 57 Possivelmente familiar Não relatado Polimorfismo do códon 210 do gene PRPN Alemanha, residente no Brasil Branca 2001 Nitrini et.al (15) Homem 40 Possivelmente familiar Não relatado Mutação no Códon 183 Não informado Não informado 2001 Nitrini et.al (16) Homem 47 Possivelmente familiar Não relatado Mutações nos nucleotídeos 321-340 Não informado Branca 1997 Nitrini et.al (17) Homem 53 Possivelmente esporádica Não relatado Não encontrado Não informado Branca 2005 Pimentel et.al (18) Homem 75 Possivelmente esporádica Não relatado Não encontrado Não informado Não informado 2022 Sales et.al (19) Homem 41 Iatrogênica Não relatado Não encontrado Não informado Não informado 2002 Smid et.al (20) Homem 59 Possivelmente familiar Mercador Mutações nos códons 200 e 129 Não informado Branca 2007 Tavares-Júnio et.al (21) Mulher 53 Possivelmente esporádica Não relatado Não encontrado Não informado Não informado 2022
Fonte: Elaborado pelos autores, 2023
Após a extração dos dados encontrados nos relatos de casos no Brasil, foi realizada a pesquisa acerca da mortalidade do CID A81, permitindo a descrição quanto a faixa etária, sexo, raça e região geográfica de maior ocorrência.
Quanto à faixa etária, o total de óbitos tende ao pico na faixa de 60 a 69 anos (GRÁFICO 1), indicando também um pico importante de incidência nessa faixa etária, sendo marcante o aumento do número de óbitos confirmados a partir da instituição da notificação compulsória.
Gráfico 1: Total de óbitos de pacientes no Brasil pelo CID A81 estratificado
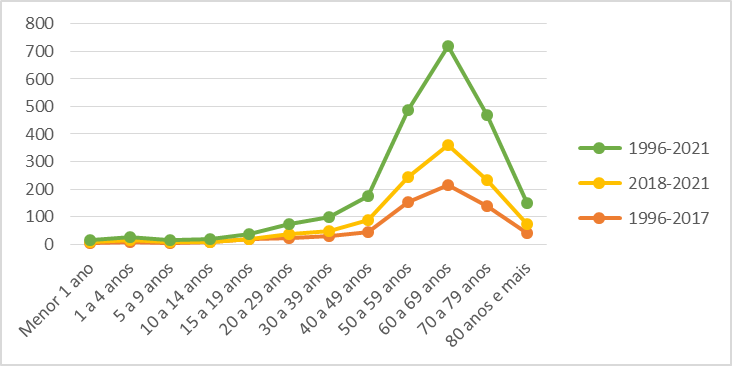
Fonte: Sistema de Informação de Mortalidade (SIM), 2023
Quanto ao sexo, o total de óbitos pende ao sexo feminino, permanecendo com um maior número de óbitos tanto de 1996-2017, quanto 2018-2021 e no período de geral de 1996-2021, no qual o sexo feminino é responsável por 52,2% do total de casos de 1996-2021 (TABELA 2).
Tabela 2: Total de óbitos de pacientes no Brasil pelo CID A81.
Sexo 1996-2017 2018-2021 1996-2021 Masculino 329 (47,81%) 215 (47,67%) 544 (47,8%) Feminino 359 (52,18%) 236 (52,32%) 595 (52,2%) Total 688 (100%) 451 (100%) 1139 (100%)
Fonte: Sistema de Informação de Mortalidade (SIM), 2023
Quanto a raça, observa-se uma predominância na população branca seguida pela população parda (TABELA 3). O número total na população branca é superior às outras populações, por exemplo o número é 16 vezes maior do que o da população negra e 3 vezes maior que na raça parda, indicando a etnia branca ser um possível fator de risco, devido ao maior número de casos presente neste grupo.
Tabela 3: Total de óbitos de pacientes no Brasil pelo CID A81 estratificado por raça nos períodos de 1996-2017, 2018-2021 e 1996-2021.
Raça 1996-2017 2018-2021 1996-2021 Branca 494 (71,8%) 272 (60,31%) 766 (67,25%) Preta 20 (2,90%) 26 (5,76%) 46 (4,03%) Amarela 10 (1,45%) 7 (1,55%) 17 (1,49%) Parda 114 (16,56%) 130 (28,82%) 244 (21,42%) Indígena 0 (0%) 2 (0,44%) 2 (0,17%) Ignorado 50 (7,26%) 14 (3,10%) 64 (5,61%) Total 688 (100%) 451 (100%) 1139 (100%)
Fonte: Sistema de Informação de Mortalidade (SIM), 2023
Quanto à região do país mais afetada, o Sudeste é a mais comum, seguido pela região Sul e Nordeste (TABELA 4). Sendo que a região Sudeste contabiliza em torno de 50% dos casos da doença
Tabela 4: Total de óbitos de pacientes no Brasil pelo CID A81 estratificado
Região 1996-2017 2018-2021 1996-2021 Região Norte 14 (2,03%) 10 (2,21%) 24 (2,10%) Região Nordeste 109 (15,84%) 117 (25,94%) 226 (19,84%) Região Sudeste 368 (53,48%) 204 (45,23%) 572 (50,21%) Região Sul 155 (22,52%) 81 (17,96%) 236 (20,71%) Região Centro-Oeste 42 (6,10%) 39 (8,64%) 81 (7,11%) Total 688 (100%) 451 (100%) 1139 (100%)
Fonte: Sistema de Informação de Mortalidade (SIM), 2023
Diante dessa perspectiva, é importante destacar que ocorre um número mais acentuado de casos no Estado de São Paulo, tendo 3 vezes mais casos que o segundo estado com maior número de casos, o Rio de Janeiro. Assim como é notável que os 3 estados com maiores números de casos (São Paulo, Rio de Janeiro e Minas Gerais) se localizam na região sudeste, solidificando a perspectiva de que a região Sudeste é o centro de casos no Brasil. (GRÁFICO 2).
Gráfico 2: Total de óbitos de paciente no Brasil pelo CID A81 estratificado por Unidade Federativa nos períodos de 1996-2017. 2018-2021 e 1996-2021
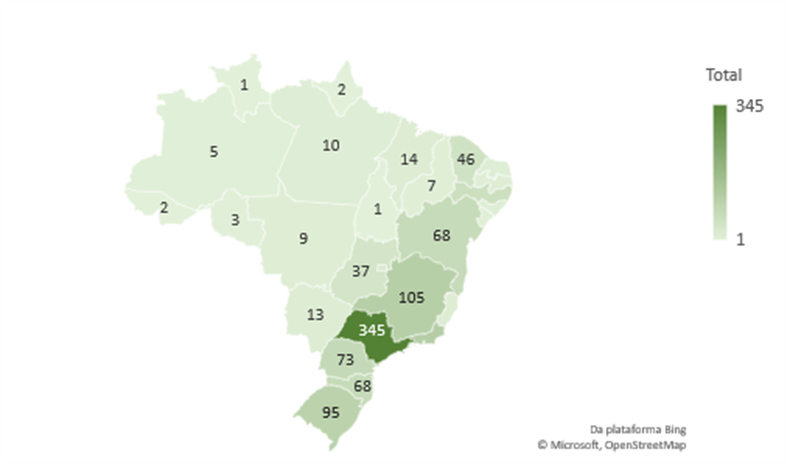
Fonte: Sistema de Informação de Mortalidade (SIM),2023
5. DISCUSSÃO
O perfil de risco para a Doença de Creutzfeldt-Jakob é consagrado na literatura como pessoas do sexo masculino com média de idade de 67 anos para a forma esporádica (1). Entretanto o perfil encontrado através dos dados do DataSUS é diferente, embora confirme a faixa etária mais comum, a população mais afetada pelo CID A81 foi a feminina, apesar dessa divergência, é importante levar em conta que a pesquisa na base do DataSUS baseou-se no CID A81 que compreende múltiplas doenças, dentre elas a CJD, a Panencefalite esclerosante subaguda, Leucoencefalopatia multifocal progressiva, assim como outras infecções por vírus atípicos do sistema nervoso central e infecção não especificada do sistema nervoso central por vírus atípicos; diante dessa miríade de doenças, é possível que a maior incidência no sexo feminino em algumas dessas outras doenças, que não são a CJD, cause essa divergência. Importante ressaltar que essa divergência não é observada nos relatos de casos, nos quais há mais relatos na população masculina, reforçando a hipótese de que há uma influência dessas outras doenças sobre os dados secundários do DataSUS.
Por outro lado, os dados do DataSUS corroboram com a literatura, visto que os casos notificados têm um pico na faixa de 60-69 anos, importante destacar que um perfil semelhante foi encontrado por Cardoso et al. (22) em um estudo baseado em dados secundários. Entretanto, quando analisamos a idade dos relatos de caso, é observável idades inferiores ao que é esperado para a doença priônica. Os motivos dessa divergência permanecem debatíveis, os dados encontrados nos relatos de caso podem indicar que no Brasil há uma tendência aos pacientes serem mais novos. Entretanto, não é absurdo considerar um viés de publicação para casos de indivíduos mais novos, tendo em vista ser de maior raridade, sendo por isso encontrado esse perfil mais jovem, se comparado ao estudo de Cardoso et al. (22) e aos dados do DataSUS, que se assemelham ao preconizado pela literatura.
Os relatos de casos ao descrever a forma da CJD muitas vezes não traziam explícito ou com certeza da forma que ali havia sido identificada, essa dificuldade no diagnóstico é associada a países de baixa e média renda nos quais há falta de laboratórios e infraestrutura adequada para o manejo e identificação de príons (3). Tais dificuldades e limitações já haviam sido relatadas em um estudo prévio no Brasil, no qual foi possível indicar que a falta de comunicação entre a vigilância sanitária e pacientes, o descuido de médicos e familiares, necrópsia não-compulsória, baixo números de profissionais treinados para realizar a necrópsia nessas situações, a falta de laboratórios equipados de maneira adequada e o preconceito com tais doenças, prejudicam de maneira significativa o diagnóstico (23).
Uma característica marcante encontrada nos relatos é a abstinência da descrição dos hábitos de vida dos pacientes diagnosticados com CJD, isso é prejudicial para que possa ser identificados os principais fatores de riscos que estão associados no país, tendo em vista que além de fatores genéticos, diversos outros fatores de riscos já foram identificados como profissão, hábitos alimentares, dentre outros. (1,3,24,25). Demonstrando que durante o relato de casos de CJD é importante maior atenção à história psicossocial do paciente, além de achados clínicos e histopatológicos.
Por outro lado, a análise genética foi mais prevalente nos relatos de casos, sendo em sua maior parte relatado uma procura por mutação, em especial do códon 129, normalmente associado com a doença (3). Tal pesquisa é importante, porque múltiplos genótipos são associados com diferentes graus de susceptibilidade à doença (1), nos relatos de casos foi observado diversos genótipos com diferentes mutações, sendo o único que foi encontrado duas vezes foi o polimorfismo do códon 129, de modo que traçar um perfil genético desta população é inviabilizado.
Ademais a incidência na etnia branca e nas regiões de colonização europeia, em especial o Sul e Sudeste do país, regiões do país conhecidas pelos focos de imigração de italianos, alemães e outras etnias brancas (26), condizem com os achados internacionais tanto de Uttley et al (1) e de Watson et al (3); que demonstram um maior risco na população europeia e, devido ao caráter genético, seus descendentes.
Além disso é importante ressaltar o impacto da notificação compulsória: no período de 1996-2017 houve a média de 32,7 de óbitos notificados, já do período de 2018-2021 a média subiu para 150,3 óbitos notificados. Isso é importante de salientar porque o protocolo de notificação compulsória da doença Creutzfeldt-Jakob é uma medida adotada tanto no âmbito internacional em vários países quanto no Brasil para garantir a vigilância epidemiológica e o controle dessa doença (3,27).
Essa notificação compulsória é fundamental para que as autoridades de saúde possam identificar e monitorar casos da doença, avaliar sua distribuição geográfica, analisar tendências e implementar medidas de prevenção e controle adequadas. No cenário internacional, existem diretrizes definidas pela Organização Mundial da Saúde (OMS) para a notificação compulsória da doença Creutzfeldt-Jakob. Essas diretrizes visam estabelecer um fluxo consistente e padronizado de informações epidemiológicas. Dentre as etapas envolvidas no processo de notificação estão incluídas (28):
1. Suspeita clínica: O primeiro passo é a suspeita clínica da doença Creutzfeldt-Jakob, que é baseada nos sintomas característicos, como deterioração do sistema nervoso central, demência rápida e alterações comportamentais.
2. Avaliação clínica: Após a suspeita clínica, é necessário realizar uma avaliação clínica detalhada, que inclui anamnese, exame neurológico e avaliação de exames complementares, como eletroencefalograma (EEG), ressonância magnética (RM) e líquor.
3. Exame neuropatológico: Um passo crucial para o diagnóstico definitivo da doença Creutzfeldt-Jakob é a análise neuropatológica do tecido cerebral. Isso geralmente envolve a realização de uma biópsia cerebral ou, mais comumente, a autópsia do paciente após o falecimento. Durante essa análise, são utilizadas técnicas de histopatologia para identificar as características típicas da doença, como a presença de placas amiloides e a perda de neurônios.
4. Microscopia eletrônica: Além da análise histopatológica convencional, a microscopia eletrônica é frequentemente usada para confirmar o diagnóstico de Creutzfeldt-Jakob. Essa técnica permite visualizar os príons, que são agentes infecciosos e nocivos responsáveis pela doença.
No âmbito do Brasil, o protocolo de notificação compulsória segue princípios semelhantes aos internacionais, com algumas particularidades adaptadas à realidade do país. No Brasil, a notificação compulsória da doença Creutzfeldt-Jakob é realizada pelo Sistema Nacional de Vigilância Epidemiológica, que recebe as informações por meio das Secretarias Estaduais de Saúde. (29).
Quanto aos critérios diagnósticos, nos casos prováveis, o modelo internacional aponta critério de ressonância magnética cerebral típica ou positividade no teste RT-QuIC (3), ausentes no nacional.
A rede europeia de vigilância da doença de Creutzfeldt-Jakob (EuroCJD), padrão internacional para vigilância, classifica a doença em 3 subtipos: CJD esporádica, CJD hereditária e CJD adquirida, a qual abrange as formas Iatrogênicas, Kuru e a Variante. Já o protocolo de notificação e investigação da doença de Creutzfeldt-Jakob do Ministério da Saúde (30) classifica diretamente em quatro subtipos, esporádica, hereditária, iatrogênica e variante, excluindo o subtipo Kuru, raro e relatado apenas em Papua Nova Guiné (1, 3).
Ademais há diferenças nos modelos de investigação da doença, por exemplo o modelo brasileiro não contempla testes como o PMCA, não busca inserir em ensaios clínicos esses pacientes, não incentiva o aconselhamento genético dentre outras (Figura 2).
Figura 2: Modelo brasileiro e internacional de investigação da CJD
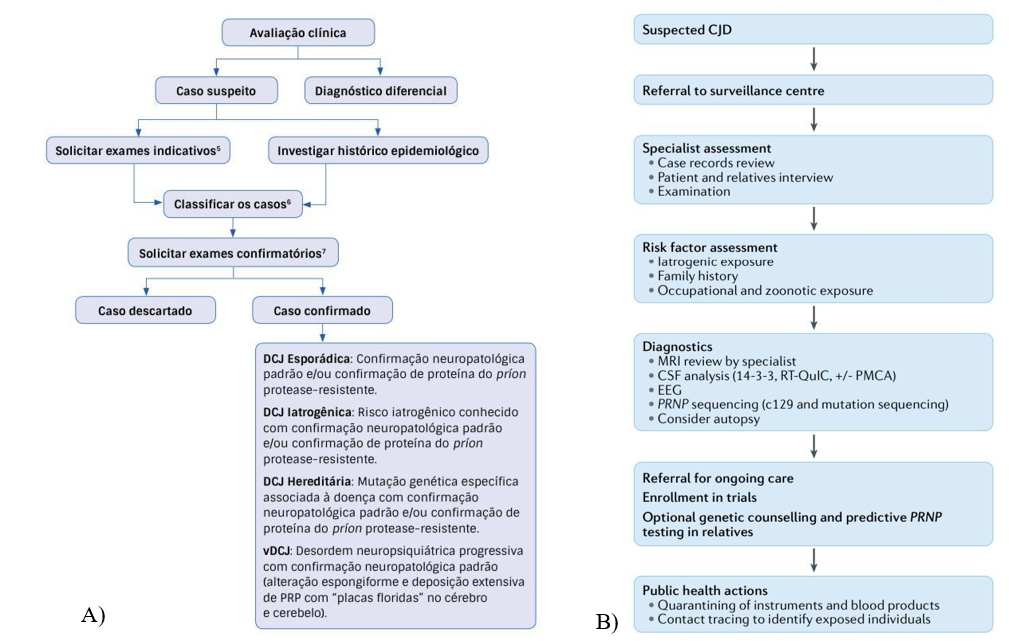
Fontes: A) Ministério da Saúde, 2018, B) Watson et al, 2021 (3). Compilado pelos autores
De acordo com os critérios internacionais, o modelo B demonstra um modelo de vigilância nacional para CJD com base em sistemas de alta fidelidade, o qual fornece uma avaliação diagnóstica detalhada, incluindo a determinação do subtipo de CJD, triagem de fatores de risco epidemiológicos importantes e avaliação e mitigação dos riscos de transmissão a outros. LCR, líquido cefalorraquidiano; PMCA, amplificação cíclica de dobramento incorreto de proteínas; RT-QuIC, conversão induzida por tremores em tempo real (31). Em contrapartida, o fluxo de investigação brasileiro não abrange alguns testes como PMCA e RT-QuIC, demonstrando a carência diagnóstica para essa rara e fatal doença em território brasileiro. Na questão da vigilância e investigação brasileira, modelo A, também não é abordado sobre inscrições em ensaios, aconselhamento genético opcional e teste PRNP preditivo em parentes e sobre rastreamento de contato para identificar indivíduos expostos. A DCJ ocorre em todo o mundo, porém em grande parte dos países continua sendo subnotificada pela carência de um sistema de vigilância adequado (3).
Os programas de vigilância em andamento para a doença de Creutzfeldt-Jakob (CJD) podem contribuir significativamente para a pesquisa da fisiopatologia da doença, testes de diagnóstico aprimorados e o desenvolvimento de tratamentos. Em primeiro lugar, esses programas fornecem uma coleta abrangente e contínua de dados sobre casos de CJD, permitindo que os pesquisadores analisem tendências, padrões e fatores de risco associados à doença. (1,3). Tais dados podem ajudar a entender os mecanismos subjacentes e a fisiopatologia da CJD. Facilitando a identificação e caracterização de diferentes subtipos de CJD, incluindo formas esporádicas, hereditárias e adquiridas.
Além disso, a vigilância contínua permite a detecção de quaisquer variantes emergentes ou formas atípicas de CJD. Ao monitorar esses casos, os pesquisadores podem investigar as possíveis rotas de transmissão, identificar novos fatores de risco e explorar as características clínicas e patológicas dessas variantes.
Os programas de vigilância desempenham um papel vital na facilitação da pesquisa sobre tratamentos potenciais. Ao identificar e monitorar os indivíduos afetados, esses programas podem ajudar no recrutamento de participantes para ensaios clínicos e estudos terapêuticos. Isso é particularmente importante considerando a natureza rapidamente progressiva e fatal da CJD, onde o diagnóstico e a intervenção precoces são cruciais. Os dados de vigilância também podem fornecer informações valiosas sobre a progressão, resultados do tratamento e potenciais alvos terapêuticos (1,3).
Por fim, esse trabalho teve algumas limitações, dentre elas: (1) Os relatos de casos carecem de todas as informações necessárias para traçar um perfil completo acerca da CJD, seja ele a partir de fatores de riscos genéticos e não-genéticos. (2) Dado a maneira com que o DataSUS disponibiliza os dados, através do CID de categoria, foi inviabilizada a pesquisa específica dos casos de CJD no sistema, de modo que os dados extraídos pelo DataSUS são apenas uma aproximação da realidade.
6. CONCLUSÃO
No Brasil, é possível afirmar que a Doença de Creutzfeldt-Jakob pode apresentar diferenças nas características da população afetada, tendo em vista as divergências encontradas ora pelos dados secundários do DataSUS e ora pelos dados encontrados nos relatos de casos. Desse modo é difícil traçar um perfil bem definido da doença com as informações atualmente disponíveis.
Sendo assim, a maneira com que se está estruturado os mecanismos de notificação e de acesso à informação precisa ser revista: a instituição da notificação compulsória da CJD foi uma medida e evolução importante para o Brasil, tendo em vista que ocorreu um aumento do número de casos identificados, entretanto, ainda é importante observar as diferenças que existem no país se comparado com os modelos internacionais, em especial na classificação da CJD e nas técnicas que são utilizadas para a detecção, dessa maneira é necessário uma atualização dos atuais sistemas e protocolos acerca da doença no Brasil, tendo em vista o papel primordial da vigilância.
Por fim a Doença de Creutzfeldt-Jakob é uma perigosa patologia de fisiopatogenia obscura, com potencial zoonótico e iatrogênico, de caráter fatal que deve estar sempre sob constante vigilância dos órgãos nacionais que devem priorizar por mecanismos de notificação e acesso à informação mais eficiente quanto a essa doença; assim como mais investimentos são necessários para sua melhor identificação e mais pesquisas devem ser realizadas tendo em vista as limitações encontradas nesse e outros estudos no Brasil.
REFERÊNCIAS.
1. Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M. Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. The Lancet Infectious Diseases [Internet]. 2020 Jan 1;20(1):e2–10. https://doi.org/10.1016/S1473-3099(19)30615-2
2. Brown P, Brandel J-P, Sato T, Nakamura Y, MacKenzie J, Will RG, et al. Iatrogenic Creutzfeldt-Jakob Disease, Final Assessment. Emerging Infectious Diseases [Internet]. 2012 Jun;18(6):901–7. https://doi.org/10.3201/eid1806.120116
3. Watson N, Brandel J-P, Green A, Hermann P, Ladogana A, Lindsay T, et al. The importance of ongoing international surveillance for Creutzfeldt–Jakob disease. Nature Reviews Neurology. 2021 May 10;17(6). https://doi.org/10.1038/s41582-021-00488-7
4. Brasil. Portaria GM/MS n 4 [Internet]. Sep 28, 2017.
5. Rodrigues B, Nakagawa F. Ministério da Agricultura confirma caso de mal da vaca louca [Internet]. CNN Brasil. CNN Brasil; 2021 [cited 2023 Nov 14]. https://www.cnnbrasil.com.br/economia/governo-do-para-confirma-caso-de-vaca-louca-no-estado (accessed 2023 Nov 14)
6. Duarte I. Ministério da Agricultura confirma que caso de vaca louca é atípico [Internet]. Estadão. 2023 [cited 2023 Nov 14]. https://www.estadao.com.br/economia/ministerio-agricultura-confirma-caso-vaca-louca-atipico (accessed 2023 Nov 14)
7. Arruda WO, Bordignon KC, Milano JB, Ramina R. Doença de Creutzfeldt-Jakob forma Heidenhain: relato de caso com achados de ressonância magnética e DWI. Arquivos de Neuro-Psiquiatria. 2004 Jun;62(2a):347–52. https://doi.org/10.1590/s0004-282×2004000200029
8. Azevedo MFA de, Nascimento F, Quintella L, Rosso ALZ de, Maranhão Filho PA, Corrêa RB, et al. Doença de Creutzfeldt-Jakob: a propósito de um caso com comprometimento medular. Arquivos de Neuro-Psiquiatria [Internet]. 2001 Dec 1;59(4):964–7. https://doi.org/10.1590/S0004-282X2001000600024
9. Brito-Marques JM de AM, Melo ES de, Medeiros FL de, Carvalho CS de, Brito-Marques PR de. Bilateral hearing loss as an initial presentation of Creutzfeldt-Jakob disease. Dementia & Neuropsychologia [Internet]. 2021 Dec 3;15(4):548–9. https://doi.org/10.1590/1980-57642021dn15-040016
10. Ciarlariello VB, Graziani O, Espay AJ, Pedroso JL. Arm Levitation as Initial Manifestation of Creutzfeldt-Jakob Disease: Case Report and Review of the Literature. PubMed [Internet]. 2018 Jan 1 [cited 2023 Nov 14];8:572–2. https://doi.org/10.7916/d80c6cgx
11. Dahy FE, Novaes CTG, Bandeira GA, Ramin LF, Oliveira ACP de, Smid J. Sporadic Creutzfeldt-Jakob disease in two clinically and virologically controlled Brazilian HIV patients who progressed rapidly to dementia: case reports and literature review. Revista do Instituto de Medicina Tropical de São Paulo [Internet]. 2021;63(23). https://doi.org/10.1590/s1678-9946202163023
12. De Souza RKM, Josviak ND, Batistela MS, Santos PSF, Landemberger MC, Ramina R. First case of V180I rare mutation in a Brazilian patient with Creutzfeldt-Jakob disease. Prion. 2017 Nov 2;11(6):465–8. https://doi.org/10.1080/19336896.2017.1397869
13. Gomes E, Matheus Ferreira Gomes, Marina de Oliveira, Niño I, Santos, Mateus Damiani Monteiro, et al. The worst is yet to come: probable sporadic Creutzfeldt–Jakob disease in a well-controlled HIV patient. Prion. 2019 Jan 1;13(1). https://doi.org/10.1080/19336896.2019.1648985
14. Huang N, Marie SKN, Kok F, Nitrini R. Familial Creutzfeldt-Jakob disease associated with a point mutation at codon 210 of the prion protein gene. Arquivos de Neuro-Psiquiatria. 2001 Dec;59(4):932–5. https://doi.org/10.1590/s0004-282×2001000600017
15. Nitrini R, Mendonça RA, Huang N, LeBlanc A, Livramento JA, Marie SK. Diffusion-weighted MRI in two cases of familial Creutzfeldt–Jakob disease. Journal of the Neurological Sciences [Internet]. 2001 Mar 1 [cited 2023 Nov 14];184(2):163–7. https://doi.org/10.1016/s0022-510x(01)00432-4
16. Nitrini R, Sérgio Rosemberg, Maria Rita Passos-Bueno, da Silva Ls, Iughetti P, Papadopoulos M, et al. Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Annals of Neurology. 1997 Aug 1;42(2):138–46. https://doi.org/10.1002/ana.410420203
17. Nitrini R, Areza-Fegyveres R, Martins VR, Maria R, Michele Christine Landemberger, Huang N, et al. Asymmetric cortical high signal on diffusion weighted-MRI in a case of Creutzfeldt-Jakob disease. Arquivos De Neuro-psiquiatria [Internet]. 2005 Jun 1 [cited 2023 Nov 14];63(2b):519–22. https://doi.org/10.1590/s0004-282×2005000300028
18. Pimentel GA, Guimarães TG, Silva GD, Scaff M. Case Report: Neurodegenerative Diseases After Severe Acute Respiratory Syndrome Coronavirus 2 Infection, a Report of Three Cases: Creutzfeldt–Jakob Disease, Rapidly Progressive Alzheimer’s Disease, and Frontotemporal Dementia. Frontiers in Neurology. 2022 Feb 7;13(1). https://doi.org/10.3389/fneur.2022.731369
19. Sales O, Huang N, Guilherme Lepski, José Antônio Livramento, Carlos Alberto Buchpiguel, Cláudia Sellitto Porto, et al. Iatrogenic Creutzfeldt-Jakob disease following human growth hormone therapy: case report. Arquivos De Neuro-psiquiatria. 2002 Jun 1;60(2B):458–61. https://doi.org/10.1590/s0004-282×2002000300022
20. Smid J, Martins VR, Landemberger MC, Riva D, Anghinah R, Nitrini R. Creutzfeldt-Jakob disease associated with a missense mutation at codon 200 of the prion protein gene in Brazil. Dementia & Neuropsychologia [Internet]. 2007 Jun [cited 2023 Nov 14];1(2):222–4. https://doi.org/10.1590/s1980-57642008dn10200017
21. Tavares-Júnior JWL, Carvalho R de O, Feitosa RRP, Rolim F de PS, Rocha FA, Pitombeira MS, et al. Diagnostic approach in a patient with Creutzfeldt-Jakob disease. Dementia & Neuropsychologia. 2022 Sep;16(3):361–4. https://doi.org/10.1590/1980-5764-dn-2021-0110
22. Cardoso CA de O, Navarro MBM de A, Soares BEC, Cardoso TA de O. Avaliação epidemiológica dos óbitos por doenças priônicas no Brasil sob o enfoque da biossegurança. Cadernos Saúde Coletiva [Internet]. 2015 Mar [cited 2020 Dec 10];23(1):2–10. https://doi.org/10.1590/1414-462×201500010002
23. Martins VR, Gomes HR, Chimelli L, Rosemberg S, Landemberger MC. Prion diseases are under compulsory notification in Brazil: Surveillance of cases evaluated by biochemical and/or genetic markers from 2005 to 2007. Dementia & Neuropsychologia [Internet]. 2007 [cited 2023 Nov 14];1(4):347–55. https://doi.org/10.1590/S1980-57642008DN10400004
24. Davanipour Z. Dietary Risk Factors for Sporadic Creutzfeldt-Jakob Disease: A Confirmatory Case-Control Study. British Journal of Medicine and Medical Research. 2014 Jan 10;4(12):2388–417. https://doi.org/10.9734/bjmmr/2014/7209
25. Hermann P, Treig J, Unkel S, Goebel S, Bunck T, Jünemann M, et al. Sporadic Creutzfeldt-Jakob Disease among Physicians, Germany, 1993–2018. Emerging Infectious Diseases. 2020 Aug;26(8). https://doi.org/10.3201/eid2608.191159
26. Mimesse E. Imigrantes europeus na América do Sul. Acta Scientiarum Human and Social Sciences [Internet]. 2014 Aug 22 [cited 2023 Nov 14];36(1):109. https://doi.org/10.4025/actascihumansoc.v36i1.21023
27. Rhoads DD, Wrona A, Foutz A, Blevins J, Glisic K, Person M, et al. Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology. 2020 Jun 22;95(8):e1017–26. https://doi.org/10.1212/wnl.0000000000010086
28. Organization WH. WHO manual for surveillance of human transmissible spongiform encephalopathies, including variant Creutzfeldt-Jakob disease [Internet]. iris.who.int. World Health Organization; 2003 [cited 2023 Nov 14].
29. WHO Guidelines on Transmissible Spongiform Encephalopathies in relation to Biological and Pharmaceutical Products World Health Organization Geneva [Internet]. 2003.
30. Biblioteca Virtual em Saúde MS [Internet]. [cited 14 nov 2023]. https://bvsms.saude.gov.br/bvs/publicacoes/protocolo_notificacao_investigacao_doenca_creutzfeldt_jakob.pdf 31. Barbosa BJAP, Castrillo BB, Alvim RP, de Brito MH, Gomes HR, Brucki SMD, et al. Second-Generation RT-QuIC Assay for the Diagnosis of Creutzfeldt-Jakob Disease Patients in Brazil. Frontiers in Bioengineering and Biotechnology [Internet]. 2020 Aug 6 [cited 2023 Nov 14];8(1). https://doi.org/10.3389/fbioe.2020.00929
1Estudante da graduação-medicina, Universidade Federal do Amazonas-UFAM. Manaus, Amazonas, Brasil. Email: viniciusbacelarferreira@gmail.com
2Estudante da graduação-medicina, Universidade Federal do Amazonas-UFAM. Manaus, Amazonas, Brasil.
3Estudante da graduação-medicina, Universidade Federal do Amazonas-UFAM. Manaus, Amazonas, Brasil.
4Estudante da graduação-medicina, Universidade Federal do Amazonas-UFAM. Manaus, Amazonas, Brasil.
5Estudante da graduação-medicina, Universidade Federal do Amazonas-UFAM. Manaus, Amazonas, Brasil.
6Doutor em Ciências da Saúde, Professor adjunto da Universidade Federal do Amazonas-UFAM, Departamento de Saúde Coletiva da Faculdade de Medicina, Manaus, Amazonas, Brasil.