DIAGNOSIS AND TESTS USED TO IDENTIFY FANCONI ANEMIA
REGISTRO DOI: 10.5281/zenodo.11068408
Layane Costa Meira1
Orientador: Alberto da Silva Santos2
RESUMO:
A Anemia de Fanconi (AF) é uma condição genética rara e progressiva caracterizada pela fragilidade cromossômica, hereditária de forma autossômica recessiva ligada ao cromossomo X. O diagnóstico desempenha um papel crucial no tratamento eficaz dos pacientes, especialmente ao considerar a maior toxicidade que os indivíduos com AF podem enfrentar com doses padrão de regimes quimioterápicos pré-transplante de células-tronco hematopoiéticas. Este estudo visa investigar e analisar a AF, uma doença genética progressiva que afeta predominantemente crianças e adolescentes. Serão incluídos os principais aspectos clínicos e moleculares, destacando os desafios associados ao diagnóstico precoce e preciso. A metodologia de pesquisa adotada consiste em uma revisão bibliográfica. Os resultados obtidos fornecem percepções importantes sobre a AF, contribuindo para a compreensão da doença e destacando a necessidade de abordagens diagnósticas mais eficazes e intervenções terapêuticas específicas. Portanto, a busca incessante por métodos diagnósticos mais eficazes, aliada a uma compreensão mais profunda da heterogeneidade genética e fenotípica da AF, são cruciais para aprimorar a qualidade de vida e a sobrevida dos pacientes.
Palavras-chaves: Anemia de fanconi, transplante de células-tronco hematopoéticas, condição genética rara.
ABSTRACT
Fanconi Anemia (FA) is a rare and progressive genetic condition caracterizem by chromosomal fragility, inherited in an autosomal recessive manner linked to the X chromosome. Diagnosis plays a crucial role in the effective treatment of patients, especially when considering the greater toxicity that individuals Patients with AF can cope with standard doses of pre-hematopoietic stem cell transplant chemotherapy regimens. This study aims to investigate and analyze FA, a progressive genetic disease that predominantly affects children and adolescents. The main clinical and molecular aspects will be included, highlighting the challenges associated with early and accurate diagnosis. The research methodology adopted consists of a bibliographic review. The results obtained provide important insights into SCA, contributing to the understanding of the disease and highlighting the need for more effective diagnostic approaches and specific therapeutic interventions. Therefore, the relentless search for more effective diagnostic methods, combined with a deeper understanding of the genetic and phenotypic heterogeneity of SCA, are crucial to improving patients’ quality of life and survival.
Key words: Fanconi anemia, hematopoietic stem cell transplantation, rare genétic condition.
INTRODUÇÃO
A Anemia de Fanconi (AF) é uma condição genética rara caracterizada pela fragilidade cromossômica, sendo hereditária de forma autossômica recessiva ligada ao cromossomo X. Esta condição afeta indivíduos de diversos grupos étnicos e é observada em aproximadamente 1 a cada 360 mil nascimentos. Pacientes com AF usualmente apresentam também alterações nos parâmetros de crescimento, com altura, peso e/ou perímetro cefálico abaixo do percentil cinco 1 -2- 3.
Esta doença é altamente heterogênea, resultando em uma ampla variedade de anomalias nos portadores, além de aumentar o risco de desenvolvimento de leucemia, displasia de medula, tumores hepáticos e carcinomas 4.
Caracteristicamente, a insuficiência medular tem progressão de forma insidiosa, levando a pancitopenia. A anemia geralmente é a última citopenia a se manifestar, o que complica o diagnóstico devido ao próprio nome da doença. Além disso, a variabilidade genotípica da doença também aumenta a dificuldade diagnostica 4.
Atualmente, o transplante de células-tronco hematopoiéticas (TCTH) representa a única abordagem terapêutica com potencial de cura hematológica para a AF. Destaca- se a necessidade crucial de identificar pacientes, considerando a escassez de estudo em nosso país que enfocam principalmente seus aspectos clínicos5-6.
O diagnóstico desempenha uma função essencial no efetivo tratamento dos pacientes. Um exemplo ilustrativo é observado em indivíduos diagnosticados com AF, que podem enfrentar maior toxicidade quando submetidos à dose padrão de regimes quimioterápicos pré-transplante de células-tronco hematopoiéticas. Dado que no início da pancitopenia, é mais tardio, é recomendável considerar o diagnóstico de AF em todas as crianças com características dismórficas distintas, menos na ausência de alterações hematológicas 7-8-9.
O objetivo deste trabalho é analisar a AF onde são citados os principais aspectos clínicos e moleculares dessa doença, bem como os desafios associados ao seu diagnóstico precoce e preciso. Por meio de uma revisão abrangente da literatura científica e da leitura de estudos clínicos, buscou-se compreender os métodos diagnósticos, como exames laboratoriais e testes genéticos, e avaliando sua eficácia na identificação precoce da AF. Além disso, abordou-se as perspectivas futuras para o diagnóstico da doença, como o uso de biomarcadores e técnicas de sequenciamento genético de última geração, com objetivo de melhorar o prognóstico e o manejo clínico dos pacientes afetados.
METODOLOGIA
Neste estudo, a metodologia de pesquisa adotada para investigar a anemia de Fanconi envolveu uma revisão bibliográfica. A pesquisa foi conduzida em várias bases de dados científicas, incluindo PubMed, Scopus, Scielo, BVS, Google acadêmico, utilizando termos de busca relevantes, como “Anemia de Fanconi, Anemia Aplásia, pancitopenia, transplante célula tronco ”. Foram considerados estudos com foco em pesquisas clínicas, revisões sistemáticas, estudos de validação de testes diagnósticos e estudos que abordam avanços recentes no diagnóstico da AF.
A seleção dos estudos foi realizada com base em critérios de inclusão e exclusão predefinidos, e a qualidade metodológica dos estudos selecionados foi avaliada de acordo com diretrizes estabelecidas. A síntese dos dados obtidos foi realizada por meio de uma análise descritiva e crítica, destacando os principais achados e lacunas de conhecimento identificados na literatura.
CONSIDERAÇÕES GERAIS
Definição da doença, epidemiologia e fatores de risco associados.
A AF é uma doença extremamente heterogênea, apresentando uma ampla gama de anormalidades. As mutações que ocorrem nos genes dos pacientes com AF impedem o reparo adequado do DNA (ácido desoxirribonucleico), ativando a apoptose celular e resultando na depleção das células-tronco hematopoiéticas, causando pancitopenia. Além disso, é bem conhecido a presença de malformações da pele (manchas café-com- leite), do sistema esquelético (alterações de polegares e rádio) (Figura 1), do sistema nervoso central e do trato geniturinário, entre outros 10-11-12.

Figura 1 – Ausência do primeiro quirodáctilo direito10
A AF apresenta uma incidência de aproximadamente 1/200.000 – 400.000, com uma prevalência mais elevada em grupos populacionais específicos, como os Judeus Asquenazes(1:30.000) e os Africânderes (1:22.000). A frequência de portadores é de 1/300 em europeuse americanos, mesmo considerando a raridade da doença 13.
No Brasil, há escassez de dados epidemiológicos que evidenciam a incidência da doença, sendo que apenas um estudo com 255 pacientes, provenientes de todas as regiões do país, revelou que 66 (25,88%) dos casos originaram-se da região nordeste. A prevalência da doença entre os sexos é contraditória, não havendo consenso. Apesar disso, a expectativa de vida tem melhorado com diagnóstico precoce e tratamentos multidisciplinares, incluindo o transplante da Medula óssea (MO). Atualmente, a expectativa de vida de um paciente com AF é de 33 anos14.
O transplante alogênico da MO permanece como a única alternativa curativa para pacientes com anemia de Fanconi. No entanto, essa abordagem enfrenta desafios devido à sensibilidade das células desses pacientes a agentes alquilantes e radiação. Portanto, é necessário submeter esses pacientes a um regime.
Os riscos associados ao pós-transplante de medula óssea (TMO) em pacientes com AF incluem a doença do enxerto contra o hospedeiro (GVHD), suscetibilidade a tumores sólidos e o aumento a infecções. Além disso, há uma probabilidade de 50% de recorrência da doença mesmo após o transplante da MO14-15.
Quadro clínico e sintomas da anemia de Fanconi:
O quadro clínico da AF fundamenta-se no histórico familiar, na presença de consanguinidade e nas características clínicas do paciente. No entanto a variabilidade fenotípica específica associada a AF torna sua solução desafiadora, uma vez que alguns indivíduos podem não manifestar sinais e sintomas, ou apresentá-los com diferentes níveis de gravidade. Esta variação pode ocorrer mesmo entre pessoas aparentadas, incluindo irmãos gêmeos 16.
Outro aspecto crucial da AF é a baixa expectativa de vida dos afetados, que, em média, é de 20 anos, sendo a probabilidade de sobrevivida acima dos 50 anos 16-17. Essa redução na expectativa de vida é atribuída principalmente a Anemia Aplásica, e a maior propensão desses pacientes ao desenvolvimento da Leucemia Mieloide Aguda (LMA), Síndrome Mielodisplásica(SMD) ou tumores sólidos, especialmente a partir dos 20 anos de idade 18-17-20.
Apesar dos avanços no tratamento das afecções hematológicas associadas a síndrome, as infecções emergem como a principal complicação clínica, mesmo com o desenvolvimento de novos antibióticos. Muitas crianças com AF acabam perdendo a vida devido a infecção bacteriana e fúngicas, sendo que as infecções neutropênicas são geralmente pouco tolerada se geralmente não respondem apenas as bactérias21.
O diagnóstico da AF pode ser facilitado quando a suspeita é levantada através da presença das anormalidades físicas, porém, nos 30% dos casos em que não ocorrem tais anormalidades é comum que o diagnóstico seja tardio, com a chegada da juventude e vida adulta. Nestes casos, os primeiros sinais são as elevações nos níveis de hemoglobina fetal emacrocitose, por isso deve-se pensar em AF quando houver casos inexplicável de macrocitose, hipoplasia, Anemia Aplásica, citopenia, SMD e LMA em crianças e jovens 22-23.
A heterogeneidade do perfil clínico dos pacientes com AF são explicados pela grande quantidade de genes, mutações e interações que são observadas no estudo genético da doença, porém esta expressão fenotípica nem sempre estar presentes nos pacientes com AF. Por isso é comum que o diagnóstico desta doença seja realizado tardiamente após a expressão das alterações hematológicas 24.
Testes genéticos e triagem da anemia de Fanconi:
O método considerado como padrão para diagnostico da AF é o teste de Quebras Cromossômicas. Este teste baseia-se na maior sensibilidade dos portadores da doença quando expostos a agentes como MMC (Mitomicina C) e DEB (Diepoxicamptotecina). Atualmente, uma análise citogénetica para detectar a instabilidade cromossômica causada por DEB é considerada o teste padrão-ouro para o diagnóstico da AF 25-26.
No entanto, é crucial ter em mente que os indivíduos heterozigotos para a AF não podem ser identificados por meio de testes com DEB/MMC. Isso ocorre porque os portadores heterozigotos são assintomáticos, e suas células não apresentam sensibilidades a fatores clastogênico, o que dificulta o diagnóstico. Apesar do teste DEB não detectar heterozigotos e gerar resultados falso-negativos, ele ainda é amplamente utilizado como principal método no diagnóstico da AF 25-26.
Alguns autores questionaram a efetividade do teste, uma vez que casos apresentados pela análise molecular podem apresentar resultados negativos no exame citogenético. Além disso, pacientes com mosaicismo na AF (geralmente uma normal e outra alterada para a síndrome)podem ter resultados falso-negativo no teste 27.
Portanto, em situações em que a análise sanguínea é normal e persiste uma incerteza diagnóstica, é crucial avaliar outro tecido com indução de quebra cromossômica, normalmente utilizado fibroblasto. Dessa forma, não se pode descartar a possibilidade de um paciente ter resultado negativo para a AF e mesmo assim ainda poder apresentar uma síndrome 28.
Um teste relacionado a AF é o teste de complementação, que envolve uma análise do fenótipo celular. A complementação é definida como a capacidade das células de pacientes com fenótipos semelhantes, devido a mutações em genes diferentes, que restabelecem o fenótipo selvagem quando hibridizados. No contexto da avaliação da AF, o fenótipo celular desenvolvido é uma instabilidade em cultura exposta a substâncias como DEB e MMC 29.
No entanto, coloca-se em dúvida a eficácia do teste, uma vez que o exame negativo é apresentado em alguns casos pela análise molecular. Além disso, pacientes com mosaicismo para fibrose cística (que possuem uma constituição genética com mais de um tipo de linhagem celular, geralmente uma normal e outra alterada para a síndrome) podem apresentar resultadofalso-negativo30.
Assim, em situações em que a análise do sangue foi normal e persiste uma incerteza diagnóstica, torna-se crucial avaliar outros tecidos com indução de quebras, geralmente fibroblastos. Dessa forma, não se pode descartar a possibilidade de alguns pacientes com resultado negativo para fibrose dística na presente amostra ainda pode apresentar a síndrome 18-31.
O diagnóstico diferencial da AF deve incluir a associação VATER/VACTERL, além da síndrome de Holt-Oram (OMIM 142900) e a síndrome de trombocitopenia e agenesia radial (TAR) (OMIM 274000)(1) devido, principalmente, às anormalidades radiais. Outras condições a serem consideradas incluem a anemia de Diamond- Blackfan (OMIM 105650) (especialmente pelo quadro hematológico), a neurofibromatose tipo 1 (OMIM 162200) (pelas manchas café-com-leite) e outras Síndromes de instabilidade cromossômica, como a síndrome de Bloom (OMIM 210900) e a síndrome de ataxia- telangiectasia (OMIM 208900).Entretanto, nenhuma dessas síndromes de instabilidade apresenta alteração no cariótipo, quando este é processado em presença de DEB/MMC 1-32-33.
Outros exames complementares relevantes.
Dada a instabilidade da transparência clínica no diagnóstico da AF, tornou-se evidente a necessidade de realizar testes laboratoriais, como hematológicos e citogenéticos, entre outros, para confirmação ou descarta do diagnóstico. Recomenda-se que esses exames sejam indicados para todas as crianças, jovens e adultos que apresentam hipoplasia, Anemia Aplástica, citopenia, macrocitose sem causa conhecida, SMD, LMA e anormalidades físicas indicativas de AF 34.
As primeiras manifestações hematológicas incluem macrocitose com poiquilocitose, anisocitose moderada, aumento do antígeno eritrocitário, persistência de hemoglobina (HB) fetal decorrente da tensão na eritropoiese e elevação da eritropoietina sérica. Os primeiros sintomas clínicos observados são petéquias e hematomas, seguidos por palidez, fadiga e suscetibilidade a infecções 35.
No entanto, os aspectos hematológicos por si só não são conclusivos para um diagnóstico preciso para a AF, uma vez que outros tipos de anemias podem compartilhar características hematológicas semelhantes. Diante desse critério, análises mais aprofundadas, como avaliações citogénetica, foram elaboradas para possibilitar uma caracterização mais precisa dos pacientes com AF 36.
Dada a extensa variabilidade genética representada por três grupos de complementação, cada um contendo genes com diversas mutações, a oferta de um diagnóstico molecular rápido se torna impraticável. No entanto, é imperativa a busca por um diagnóstico preciso e oportuno, dado o desenvolvimento precoce da doença em direção a malignidade e a urgência em encontrar doadores compatíveis para futuro transplantes de células-tronco hematopoiéticas37.
A sensibilidade acentuada das células AF ao efeito clastogênico, especificamente à quebra de cromossomos provocados por agentes de reticulação de DNA, como o DEB, serve como um marcador distintivo crucial para o diagnóstico37.
Em estudos adicionais, foi estabelecida uma metodologia para diagnóstico laboratorial da AF com base na notável sensibilidade das células afetadas pela AF a ação de agentes clastogênicos que induzem a formações cruzadas (Figura 2). Observou-se um aumento no número de aberrações cromossômicas em células com AF quando expostos a esses agentes clastogênico. A inclusão dessas substâncias no cultivo celular, devido a sua capacidade de facilitar o diagnóstico, tornou-se o método padrão 37.
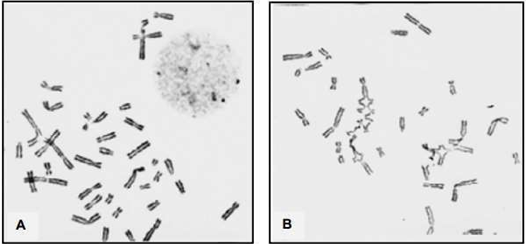
Figura 2 – Metáfases de pacientes portadores de AF representando alterações cromossômicas espontâneas (A) e induzidas por exposição ao DEB (B) em células linfocitárias37.
CONSIDERAÇÕES FINAIS
Em resumo, a AF surge como uma entidade clínica desafiadora e multifacetada, exigindo uma abordagem abrangente por partes dos profissionais de saúde no processo diagnóstico e terapêuticos. As mutações genéticas subjacentes não comprometem apenas a hematopoese, mas também desencadeiam uma cascata de efeitos adversos que reverberam em vários sistemas orgânicos.
Embora persista uma lacuna de dados epidemiológicos no Brasil, estudos indicam uma concentração notável de casos na região nordeste, sublinhando a necessidade de maior atenção a essa condição no âmbito nacional. O avanço nas abordagens terapêuticas, particularmente com o diagnóstico precoce e tratamentos multidisciplinares, como o transplante da MO, contribuiu para uma melhoria significativa na expectativa de vida dos pacientes.
Contudo, não podemos ignorar os desafios pós-transplante, incluindo a ameaça da doença do enxerto contra o hospedeiro e a possibilidade de recorrência da AF. O quadro clínico heterogêneo da AF, muitas vezes carente de anormalidades físicas evidentes, complica o diagnóstico, enfatizando a importância de testes genéticos, como o teste de Quebras Cromossômicas, mesmo diante da complexidade de identificação de portadores heterozigotos.
Embora o transplante da MO permaneça como a única opção curativa, sua eficiência é inevitável, a despeito da sensibilidade celular dos pacientes. Os defeitos associados aos testes genéticos e de complementação também merecem atenção, destacando a necessidade de estratégias que enfrentam a variabilidade genética da AF.
A expectativa de vida amplia reflete não apenas os progressos nas terapias existentes, mas também aponta para uma direção promissória com a inclusão de novas metodologias diagnósticas, como os testes de complementação. Essas abordagens oferecem uma perspectiva encorajadora para a identificação mais precisa de casos, especialmente quando a expressão fenotípica é sutil.
Diante disso, a busca incessante por métodos diagnósticos mais eficazes, aliada a uma compreensão mais profunda da heterogeneidade genética e fenotípica da AF, emerge como uma necessidade premente para aprimorar a qualidade de vida e a sobrevida dos pacientes. A AF transcende o escopo de um desafio clínico, sendo uma oportunidade excelente para a comunidade científica avançar na compreensão das intrincadas relações entre genética, hematologia e tratamento de doenças raras.
REFERÊNCIAS
1. Sagaseta IM, Molina J, Lezáun I, Valiente A, Durán G. Anemia de Fanconi. Consideraciones actuales. Anales Sis San Navarra, 2003, Vol.23.
2. Keneddy RD, D’Andrea AD. DNA Repair Pathways in Clinical Practice: Lessons From Pediatric Câncer Susceptibility Syndromes, 2006, vol. 24.
3. Auerbach AD, Adler B, Chaganti RS. Prenatal and Postnatal Diagnoses and Carrier Detection of Fanconi anemia by a Cytogenetic method. Pediatrics ,1981.
4. Tootian S, Mahjoubi F, Rahnama M, Hormozian F, Mortezapour F, Razazian F et al Cytogenetic investigation in Iranian patients suspected with Fanconi anemia. J Pediatr Hematol Oncol, p.28:834-6,2006.
5. Buchwald M, Carreau M. Getic basis of Fanconi´s aemia. In: Schrezenmeier H, Bacigalupo A, editors. Aplastic anemia patho-physiology and treatment. Cambridge UK: Cambridge iversity Press ,338-54,2000.
6. Medeiros C, Zanis-Neto J, Pasquini R. Bone marrow transplantation for patients with Fanconi anemia: reduced doses of cyclophosphamide without irradiation as conditioning. Bone Marrow Transpl ,p. 24(8):849-52,1999.
7. Jones K. Smith’s recognizable patterns of human malformation.ed. Philadelphia: Elsevier, Rev.6 ,2006.
8. Dufour C, Svahn J. Fanconi anemia: new strategies. Bone Marrow Transplant, Rev.2, p. 90-5,2008.
9. Pinto FO, Leblanc T, Chamousset D, Le Roux G, Brethon B, Cassinat B.Diagnosis of Fanconi anemia in patients with bone marrow failure, 94:487-95,2009.
10. Kennedy RD, D’Andrea AD, J Clin Oncol. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes, 24:3799-808,2006.
11. Soulier J. Fanconi anemia. Hematology Am Soc Hematol EducProgram,1:492-497,2011.
12. Geiselhart A, Lier A, Walter D, Milsom MD. Disrupted signaling through the Fanconi Anemia pathway leads to dysfunctional hematopoietic stem cell biology: underlying mechanisms and potential therapeutic strategies,2012.
13. Hindricks G, Potpara T, Dagres N, Arbelo E, Bax JJ, Blomström-Lundqvist C, Boriani G, Castella M , Dan GA, Dilaveris PE, Fauchier L, Filippatos G, Kalman JM, La Meir M, Lane DA, Lebeau JP , Lettino M, Lip GYH, Pinto FJ, Thomas GN, Valgimigli M, Van Gelder IC, Van Putte BP, Watkins CL; Grupo de Documentos Científicos ESC ,Vol.10,2021.
14. Mamrak NE, Shimamura A, Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017.
15. Virts, E.L., et al., AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia. Hum Mol Genet,2015.
16. Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet , 2:446-57,2001.
17. Jones K. Smith’s recognizable patterns of human malformation. 6th ed. Philadelphia: Elsevier; 2006.
18. Sagaseta IM, Molina J, Lezáun I, Valiente A, Durán G. Anemia de Fanconi. Consideraciones actuales. Anales Sis San Navarra,2003, 26:63-78.
19. Zhang XX. Chromossome instability. In: Gersen SL, Keagle MB. The principles of clinical cytogenetics. 2nd ed. New Jersey: Humana Press; p. 350-1,2005.
20. Green AM, Kupfer GM. Fanconi anemia. Hematol Oncol Clin North Am , 23:193-214,2009.
21. Bagby G. C.; Alter B. P. Fanconi anemia. Seminars in Hematology, v.43, 2006.
22. Bagby G. C Sloand EM, Schiffer CA. Insuficiência Medular em Hematologia:Livro do Programa Educação da Sociedade Americana de Hematologia, 318-316,2004.
23. Dokal I. The genetics of Fanconi’s anaemia. Baillieres Best Pract Res Clin Haematol, Vol.13, n. 3, 2000.
24. MacMillan M. L, Wagner J. E. Haematopoeitic cell transplantation for Fanconi anaemia – when and how? British Journal of Haematology, Vol.149, n.1, p.14-21, 2010.
25. Sagaseta IM, Molina J, Lezáun I, Valiente A, Durán G. Anemia de Fanconi. Consideraciones actuales. Anales Sis San Navarra, 26:63-78,2003.
26. Tischkowitz MD, Hodgson SV, J Med Genet. Fanconi anaemia. 2003.
27. Chen H. Atlas of genetic diagnosis and counseling. New Jersey: Humana Press, 2006.
28. Auerbach AD, Adler B, Chaganti RS. Prenatal and postnatal diagnosis and carrier detection of Fanconi anemia by a cytogenetic method. Pediatrics, 67:128-35,1981.
29. Taniguchi T. Fanconi Anemia. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA): University of Washington, 1993.
30. Thompson M, Stephen L.R, Roger. W. Pure Appl. Chem,2002,Vol.75.
31. Lima CS, Lourenço GJ, Rodriguez DE, Zocca M, Bertuzzo CS. Cytogenetic and molecular diagnosis of Fanconi anemia. Rev Bras Hematol Hemoter , Vol.25,p.191-2,2003.
32. Taniguchi T. Fanconi Anemia. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA): University of Washington,1993.
33. Green AM, Kupfer GM. Fanconi anemia. Hematol Oncol Clin North Am ,2009.
34. Auerbachc A. D. Fanconi anemia diagnosis and diepoxibutane (DEB) test. Experimental Hematology,1993.
35. Magdalena N, Pilonetto DV, Bitencourt MA, Pereira NF, Ribeiro RC, Jeng M etal Frequency of Fanconi anemia in Brazil and efficacy of screening for the FANCA 3788- 3790del mutation. Braz J Med Biol, 2005.
36. Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutat Res ,2009.
37. Cohem M.M, Fruchtman C.E, Simpson S.J, Martin A.O. The cytogenetic response of Fanconi’s anemia lymphoblastoid cell lines to various clastogens. Cytogenetics and Cell Genetics, Vol.34, p.230-40, 1982.
1Graduanda em Biomedicina no Centro Universitário das Faculdades Metropolitanas Unidas-FMU, Av. SantoAmaro, 1239. Vila Nova Conceição – São Paulo-SP CEP: 04505-002, Brasil.
Layane2002costta@gmail.com
2Biomédico, Faculdade de tecnologia e ciências-FTC, Av. Adolfo Ribeiro,375, São Judas Tadeu, Jequié- BA,CEP:45204- 068, Brasil.