DIAGNOSIS OF RETT SYNDROME IN AN INFANT WITH NEUROPSYCHOMOTOR DEVELOPMENT REGRESSION: A CASE REPORT
REGISTRO DOI: 10.69849/revistaft/ra10202503271704
Ana Paula da Cunha Panis
Hugo Tadashi Oshiro Távora
Resumo
Este relato apresenta o caso de uma paciente do sexo feminino, lactente, diagnosticada com Síndrome de Rett após investigação de regressão do desenvolvimento neuropsicomotor (DNPM). A paciente foi encaminhada para avaliação em uma unidade de pediatria devido a uma regressão progressiva das habilidades adquiridas aos 18 meses de idade, associada a movimentos estereotipados e irritabilidade significativa. Exames complementares confirmaram o diagnóstico por meio de teste genético. Este caso destaca os desafios no reconhecimento e manejo precoce da Síndrome de Rett, enfatizando a importância de intervenções precoces e acompanhamento multidisciplinar.
Palavras-chave: Síndrome de Rett; Transtornos do Neurodesenvolvimento; Deficiências do Desenvolvimento.
1 INTRODUÇÃO
A Síndrome de Rett (SR) é uma desordem genética rara, causada por mutações no gene MECP2, localizado na região Xq28 do cromossomo X, que afeta principalmente meninas.(BARTHOLDI et al., 2006) Caracteriza-se por regressão gradual de habilidades previamente adquiridas, associada a grave deterioração do desenvolvimento neuropsicomotor. (LORENA et al., 2016) Essa enfermidade afeta aproximadamente 1 em cada 10.000 nascidos vivos do sexo feminino e é letal na grande maioria dos casos do sexo masculino. Trata-se de uma das principais síndromes de desenvolvimento conhecidas que são associadas a deficiência intelectual.(LORENA et al., 2016)
A SR foi descrita pela primeira vez em 1966 pelo médico Andreas Rett, que identificou decaimento neuromotor em crianças do sexo feminino, frequentemente associado à hiperamonemia.(SCHWARTZMAN, 2003) Contudo, a condição recebeu maior reconhecimento e compreensão após a publicação de Bengt Hagberg, que detalhou 35 casos em um estudo seminal, consolidando o termo “Síndrome de Rett“.(SCHWARTZMAN, 2003) Embora tradicionalmente se considere que meninas com SR apresentam desenvolvimento aparentemente normal até os seis a dezoito meses de idade, estudos mais recentes sugerem que sinais precoces, como atraso motor, hipotonia muscular e dificuldade para engatinhar, estão frequentemente presentes antes do início da regressão.(AMIR et al., 1999)
As manifestações clínicas da SR incluem características típicas, como perda da linguagem, disfunções motoras progressivas, escoliose, falha no crescimento, epilepsia e déficit mineral ósseo, aumentando o risco de fraturas. (QUEIROZ; BARROS; CALLES, 2014) Também são comuns anormalidades cardíacas, disfunções autonômicas (pés frios, distúrbios vasomotores), padrão respiratório desorganizado (apneia e hiperventilação) e distúrbios do sono, comprometendo a qualidade de vida. (QUEIROZ; BARROS; CALLES, 2014)
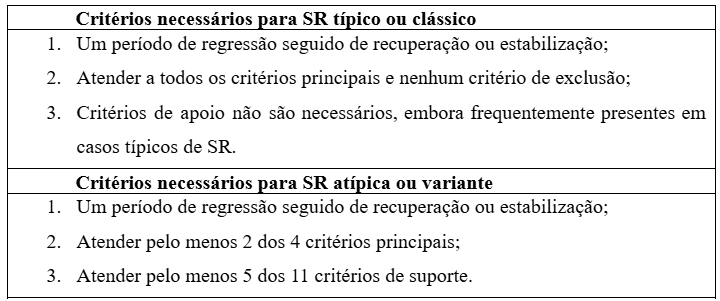
O diagnóstico da SR é baseado em uma avaliação clínica detalhada, complementada pela identificação de mutações no gene MECP2 por meio de testes genéticos. O eletroencefalograma (EEG) frequentemente apresenta anormalidades significativas; contudo, em estágios iniciais da doença, esses achados podem ser menos evidentes. (JEFFREY L. NEUL et al., 2010; PAZETO et al., 2013) A confirmação diagnóstica requer aplicação de critérios específicos, incluindo critérios principais obrigatórios, critérios de suporte e critérios de exclusão (Tabelas 1 e 2).(GOLD et al., 2024)
A SR, por ser uma doença sem cura, tem o tratamento focado no manejo dos sintomas e no suporte multidisciplinar, envolvendo uma equipe com psicoterapeutas, fisioterapeutas, fonoaudiólogos e nutricionista. (GOLD et al., 2024)
Este relato descreve o caso de uma paciente diagnosticada com a SR, ressaltando aspectos clínicos, diagnósticos e terapêuticos relevantes.
Tabela 1: Critérios diagnósticos revisados para a Síndrome de Rett (SR)

Tabela 2: Critérios necessários para identificação da Síndrome de Rett (SR) nos casos típicos ou atípicos
2 APRESENTAÇÃO DO CASO
Lactente do sexo feminino, encaminhada para avaliação devido à regressão do desenvolvimento neuropsicomotor (DNPM) observada aos 18 meses. A criança nasceu a termo, de uma gestação sem intercorrências, por parto vaginal, com peso adequado para idade gestacional e Apgar 8/9. Na triagem neonatal, identificou-se alteração no Teste da Orelhinha (o qual não foi repetido). Porém, o restante da triagem neonatal não apresentou alterações. Os pais, jovens e não consanguíneos, negam doenças genéticas ou semelhantes na família.
O desenvolvimento inicial foi considerado no limiar da normalidade. A paciente atingiu marcos como sustentação cefálica aos dois meses, sentar-se sem apoio aos oito meses e engatinhar ao redor de um ano. Contudo, aos 18 meses, ficou evidente para os pais a regressão de habilidades que já haviam sido adquiridas. A criança apresentou perda das palavras aprendidas, passando a emitir apenas vocalizações incompreensíveis. Além disso, perdeu o uso funcional das mãos e apresentou movimentos estereotipados, como levar as mãos à boca repetidamente. Manifestou um episódio de irritabilidade intensa, com choro, comportamentos de automutilação, com mordidas e arranhões, além de agressões a terceiros.
No momento da avaliação, a paciente apresentava baixo peso (9 kg, Z-score -2,2), comprimento de 81,4 cm (Z-score -1,63) e microcefalia relativa, com perímetro cefálico de 46 cm. Durante o exame físico, a criança não deambulava, mesmo com apoio, exibindo postura em abdução dos pés e pernas, base alargada, marcha com apoio em padrão equino e tônus reduzido. Demonstrou movimentos estereotipados de mãos, reflexos patelares e aquileus aumentados e não conseguia vencer a gravidade ao levantar as pernas penduladas. Não identificou-se sinais de irritação meníngea e a imunização estava atualizada. Foi internada na enfermaria para investigação diagnóstica, e solicitada avaliação com neuropediatra após suspeita de paralisia em membro inferior direito.
Após avaliação da neuropediatra, com base nos achados clínicos, incluindo regressão do DNPM, características de transtorno do espectro autista e microcefalia, a Síndrome de Rett foi considerada a principal hipótese diagnóstica. Para investigação, foram solicitados exames complementares, como tomografia computadorizada de crânio, eletroencefalograma (EEG) e ressonância magnética de crânio, todos sem alterações. Adicionalmente, foi realizado um potencial evocado auditivo e análise genética por meio do painel “Movimente/Medelics”, que identificou a presença de uma variante patogênica no gene MECP2, confirmando o diagnóstico de Síndrome de Rett.
A criança foi encaminhada para seguimento multidisciplinar com geneticista, fisioterapeuta, fonoaudiólogo e neuroestimulação.
3 DISCUSSÃO
A SR é uma rara desordem do desenvolvimento neurológico, que afeta meninas e que é normalmente é letal em indivíduos do sexo masculino. (GOLD et al., 2024) Em sua maioria, as crianças afetadas nascem sem complicações e apresentam desenvolvimento aparentemente normal até os seis meses de vida.(CABAL-HERRERA; BEATTY, 2024) No caso relatado, as primeiras alterações só foram percebidas aos 18 meses de idade, quando notou-se a regressão de habilidades psicomotoras – característica marcante na SR.
A doença evolui em quatro estágios: estagnação precoce (6-18 meses), com interrupção do desenvolvimento, isolamento e diminuição da interação social; estágio destrutivo rápido (1-3 anos), marcado por regressão psicomotora acelerada, choro injustificado, irritabilidade, perda da fala, comportamentos autistas e movimentos estereotipados das mãos; pseudo estacionário, com estabilização aparente dos sintomas, melhora parcial do contato social, mas com piora dos distúrbios motores, como ataxia, apraxia, espasticidade, escoliose e bruxismo; e deterioração motora tardia (a partir dos 10 anos), caracterizada por progressão lenta dos déficits motores, agravamento da escoliose e deficiência intelectual severa.(DUNN, 2001) Os achados do caso relatado estão em concordância com a literatura, uma vez que, no momento do diagnóstico, aos 18 meses de idade, a criança apresentava as características clínicas típicas do estágio destrutivo rápido.
Os exames de neuroimagem e o EEG são úteis para excluir condições neurológicas que podem causar sinais e sintomas semelhantes, como epilepsia, encefalopatias e doenças neurodegenerativas primárias, as quais compõem o diagnóstico diferencial da SR. O EEG desempenha um papel relevante na avaliação da SR, fornecendo informações sobre a função cortical e a gravidade clínica da doença. (SABY et al., 2024) Estudos mostram que pacientes com SR frequentemente apresentam anormalidades no EEG, como aumento da atividade de baixa frequência e redução da atividade de alta frequência. (DUNN, 2001) Entretanto, em estágios iniciais, o EEG pode ser normal, similar ao ocorrido com a paciente desse relato. Dessa forma, ressalta-se a necessidade de uma avaliação clínica detalhada e de confirmação genética para o diagnóstico.(KEOGH et al., 2018) Por fim, a identificação de uma variante patogênica no gene MECP2 foi determinante para confirmar o diagnóstico, em conformidade com as diretrizes atuais, que consideram a análise genética o padrão-ouro para a identificação da SR.(COOLEY COLEMAN et al., 2022; HUPPKE, 2000)
O manejo multidisciplinar desempenha um papel central no cuidado de pacientes com SR.(BANERJEE et al., 2019) A paciente foi encaminhada para acompanhamento com geneticista, fisioterapeuta, fonoaudiólogo e neuroestimulação, destacando a importância de integrar diferentes especialidades para otimizar o manejo. Estudos sugerem que intervenções precoces com fisioterapia e terapia ocupacional podem retardar a progressão das disfunções motoras e melhorar a qualidade de vida. (M. et al., 2019)
Não existe uma terapêutica específica ou curativa para a SR o que torna os objetivos do tratamento um tema amplamente discutido. Geralmente, as abordagens terapêuticas focam na prevenção da progressão dos sintomas, considerando a história natural da doença, ou no manejo dos sintomas já manifestados. (TOAZA et al., 2020) Recentemente, a trofinetida, um análogo sintético do tripeptídeo N-terminal do fator de crescimento semelhante à insulina I (IGF1), foi aprovada pela FDA como o primeiro agente específico para o tratamento da SR, sendo indicada para adultos e crianças a partir de 2 anos de idade.(ABO ZEID et al., 2024; BANERJEE et al., 2019) Já existem estudos em terapias gênicas, como substituição gênica e edição de RNA, que estão em desenvolvimento e representam abordagens promissoras para o manejo da síndrome.(PANAYOTIS et al., 2023)
Este caso também reforça a importância de identificar sinais preambulares da SR, que, embora sutis, orientam o diagnóstico antes da regressão mais evidente. Sabe-se que a detecção precoce está associada a um melhor planejamento terapêutico e a intervenções mais eficazes.(HUPPKE, 2000) Além disso, o relato contribui para enfatizar a necessidade de integrar o cuidado pediátrico geral ao acompanhamento especializado e garantir uma abordagem abrangente e contínua.
4 CONCLUSÃO
O caso clínico destaca a importância do acompanhamento contínuo do neurodesenvolvimento infantil para a identificação precoce de alterações, como na SR. O diagnóstico, quando feito o mais cedo possível, permite intervenções direcionadas que podem melhorar a qualidade de vida do paciente e de sua família. O relato reforça a necessidade de integrar avaliação clínica detalhada com exames complementares e adotar uma abordagem multidisciplinar no manejo, abrangendo várias áreas da saúde. Além disso, fica evidente o papel essencial da puericultura na detecção e reabilitação de distúrbios do desenvolvimento infantil.
REFERÊNCIAS
ABO ZEID, M. et al. A meta-analysis of the efficacy and safety of trofinetide in patients with rett syndrome. Neurological Sciences, v. 45, n. 10, p. 4767–4778, 21 out. 2024.
AMIR, R. E. et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics, v. 23, n. 2, p. 185–188, out. 1999.
BANERJEE, A. et al. Towards a better diagnosis and treatment of Rett syndrome: a model synaptic disorder. Brain, v. 142, n. 2, p. 239–248, 1 fev. 2019.
BARTHOLDI, D. et al. Clinical profiles of four patients with Rett syndrome carrying a novel exon 1 mutation or genomic rearrangement in the gene. Clinical Genetics, v. 69, n. 4, p. 319–326, 30 abr. 2006.
CABAL-HERRERA, A. M.; BEATTY, C. W. [Rett syndrome: from pathophysiology to developments in treatment]. Medicina, v. 84 Suppl 3, p. 45–49, set. 2024.
COOLEY COLEMAN, J. A. et al. Mosaicism of common pathogenic MECP2 variants identified in two males with a clinical diagnosis of syndrome. American Journal of Medical Genetics Part A, v. 188, n. 10, p. 2988–2998, 4 out. 2022.
DUNN, H. G. Importance of Rett syndrome in child neurology. Brain and Development, v. 23, p. S38–S43, dez. 2001.
GOLD, W. A. et al. Rett syndrome. Nature Reviews Disease Primers, v. 10, n. 1, p. 84, 7 nov. 2024.
HUPPKE, P. Rett syndrome: analysis of MECP2 and clinical characterization of 31 patients. Human Molecular Genetics, v. 9, n. 9, p. 1369–1375, 22 maio 2000.
JEFFREY L. NEUL et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Annals of Neurology, v. 68, n. 6, p. 944–950, 8 dez. 2010.
KEOGH, C. et al. Clinical and genetic Rett syndrome variants are defined by stable electrophysiological profiles. BMC Pediatrics, v. 18, n. 1, p. 333, 19 dez. 2018.
LORENA, N. et al. Rett Syndrome: a review of the literature. J Health S ed. [s.l: s.n.].
M., C. T. et al. Síndrome de Rett e Hidroterapia. Revista Neurociências, v. 12, n. 2, p. 77–81, 23 jan. 2019.
PANAYOTIS, N. et al. State‐of‐the‐art therapies for Rett syndrome. Developmental Medicine & Child Neurology, v. 65, n. 2, p. 162–170, 3 fev. 2023.
PAZETO, T. D. C. B. et al. Síndrome De Rett: Artigo De Revisão. [s.l: s.n.]. v. 13
QUEIROZ, C. M. B. DE; BARROS, J. E. S. L.; CALLES, A. C. DO N. As Características Da Síndrome De Ret T : Uma Revisão De Literatura. [s.l: s.n.]. v. 2
SABY, J. N. et al. ElectroencephalographicCorrelates of Clinical Severity in the Natural history study of RTT and Related Disorders. Annals of Neurology, v. 96, n. 1, p. 175–186, 9 jul. 2024.
SCHWARTZMAN, J. S. Síndrome de Rett. Revista Brasileira de Psiquiatria, v. 25, n. 2, p. 110–113, jun. 2003.
TOAZA, E. et al. SÍNDROME DE RETT NA ATENÇÃO PRIMÁRIA À SAÚDE: RELATO DE CASO. Revista Interdisciplinar em Ciências da Saúde e Biológicas, v. 4, n. 2, p. 47–52, 30 dez. 2020.