REGISTRO DOI: 10.5281/zenodo.7226652
Autoria de:
Carla Tamires de Souza1
Caroline Waideman Peres1
Nathaly Barbara Silva1
Sarah Witkowski Costa1
Rafael Leite Carvalho2
Priscila Ferreira Silva2
RESUMO
A síndrome de Down é uma condição na qual afeta 1 a cada 961 bebês sendo chamada de trissomia devido a uma cópia extra gerada do cromossomo 21, onde afeta diretamente no desenvolvimento cognitivo dos portadores. O cérebro possui tamanho e formato diferentes em relação ao das crianças não portadoras da síndrome, fato este que origina perfis neurocognitivos e neurocomportamentais únicos e que necessitam ser esclarecidos para um eficaz tratamento que vise a qualidade de vida das crianças que possuem esta condição. Estas tendem a ter maiores dificuldades de linguagem do que seria previsto pela idade mental, enquanto o raciocínio não verbal é uma força relativa. O funcionamento neuropsicológico da SD é caracterizado pela presença de múltiplas áreas afetadas. No entanto, os conceitos de plasticidade sugerem que podemos criar rotas neuronais e novas estratégias cognitivas que possibilitem a minimização e/ou superação dos déficits apresentados. Este estudo é importante pois nele é possível atualizar pais e profissionais que trabalham na área elucidando sobre o local afetado no sistema nervoso central, para que seja plenamente entendido a origem da condição da criança com SD, a fim de verificar tratamentos para estímulo da cognição e evolução motora da mesma. Para desenvolver esta dissertação, foi realizada uma revisão de literatura, onde a obtenção de dados ocorreu através de bancos de dados online. Após a obtenção e análise de dados, houveram resultados de 34 artigos. Concluise que indivíduos com síndrome de Down (SD) geralmente possuem perfis neurocognitivos e neurocomportamentais únicos que surgem em períodos específicos de desenvolvimento. Esses perfis são distintos em relação a outros com deficiência intelectual (DI) semelhante e refletem achados neuroanatômicos subjacentes, fornecendo suporte para um perfil fenotípico distinto.
Palavras chave: Síndrome de Down, cognição, desenvolvimento cognitivo.
ABSTRACT
Down syndrome is a condition in which it affects 1 in 961 babies being called trisomy due to an extra copy generated from chromosome 21, where it directly affects the cognitive development of carriers. The brain has a different size and shape compared to children without the syndrome, a fact that gives rise to unique neurocognitive and neurobehavioral profiles that need to be clarified for an effective treatment aimed at the quality of life of children who have this condition. These tend to have greater language difficulties than would be predicted by mental age, while non-verbal reasoning is a relative strength. The neuropsychological functioning of DS is characterized by the presence of multiple affected areas. However, the concepts of plasticity suggest that we can create neuronal routes and new cognitive strategies that make it possible to minimize and/or overcome the deficits presented. This study is important because it is possible to update parents and professionals who work in the area, elucidating the affected site in the central nervous system, so that the origin of the condition of the child with DS is fully understood, in order to verify treatments to stimulate cognition and its motor evolution. To develop this dissertation, a literature review was carried out, where data collection took place through online databases. After obtaining and analyzing data, there were results from 28 articles. It is concluded that individuals with Down syndrome (DS) generally have unique neurocognitive and neurobehavioral profiles that arise at specific periods of development. These profiles are distinct from others with similar intellectual disability (ID) and reflect underlying neuroanatomical findings, providing support for a distinct phenotypic profile.
Keywords: Down syndrome, cognition, cognitive development.
INTRODUÇÃO
A síndrome de Down (SD) é provavelmente a síndrome de identificação genética (cromossômica) mais comum, ocorrendo em aproximadamente 1 em cada 7001000 nascimentos. A SD afeta muitas áreas do desenvolvimento, principalmente a estrutura craniofacial, e causa problemas relacionados à saúde, como doenças cardíacas congênitas, anormalidades do sistema imunológico e endócrino e déficit cognitivo. É uma condição na qual uma pessoa tem um cromossomo extra. Os cromossomos são pequenos “pacotes” de genes no corpo. Eles determinam como o corpo de um bebê se forma e funciona à medida que cresce durante a gravidez e após o nascimento. Normalmente, um bebê nasce com 46 cromossomos. Bebês com síndrome de Down têm uma cópia extra de um desses cromossomos, o cromossomo 21.
Um termo médico utilizado para essa cópia extra de um cromossomo é ‘trissomia’. A síndrome de Down também é conhecida como Trissomia 21. Essa cópia extra altera a forma como o corpo e o cérebro do bebê se desenvolvem, podendo o causar desafios mentais e físicos. Mesmo que os portadores possam agir e parecer semelhantes, cada um possui habilidades diferentes. As crianças geralmente possuem um QI (uma medida de inteligência) na faixa de leve a moderadamente baixa e são mais lentas para falar do que as que possuem desenvolvimento típico (1). A síndrome ocasiona complicações clínicas, que podem interferir no desenvolvimento global da criança portadora, sendo as mais frequentes: alterações cardíacas, hipotonia, complicações respiratórias e alterações sensoriais, sobretudo relacionadas à visão e à audição. Apesar das diferentes formas de manifestação da trissomia, podem ser provocadas variações físicas, clínicas e nas capacidades cognitivas. O desenvolvimento cognitivo é um processo progressivo que pode ter validações diferentes para cada indivíduo. Dentre diversas disfunções genéticas, há uma necessidade de atualização de informações, principalmente para que os pais e responsáveis tenham conhecimento de como contribuir no crescimento e na vida social das crianças que possuem essa condição genética (2).
O cérebro de uma criança com síndrome de Down desenvolve-se diferentemente de um normal, atingindo uma forma reduzida em tamanho e alterada em configuração. Diretamente relacionadas ao retardo mental estão as modificações neuronais que se manifestam como alterações da laminação cortical, redução das ramificações dendríticas e diminuição da formação sináptica. Em contraste com crianças com desenvolvimento típico, há um declínio gradual no QI a partir do primeiro ano de vida. O declínio entre crianças com SD é aparente desde a infância e perdura até a adolescência. Esse declínio no QI reflete uma desaceleração em relação ao desenvolvimento típico, proporcionando uma deterioração no aprendizado e no desenvolvimento cognitivo.
OBJETIVOS
Gerais:
Realização do diagnóstico da síndrome de Down desde a gestação, identificação do local afetado no sistema nervoso central, análise das consequências e condução do desenvolvimento cognitivo com ênfase em aprendizado e evolução mental, além de verificar melhorias da cognição perante a plasticidade cerebral de acordo com o crescimento. Secundar profissionais no auxílio aos pais desde o diagnóstico, com tratamentos que visem o estímulo cerebral junto a melhoria da qualidade de vida e inclusão social conforme a evolução da criança.
Específicos:
- Desenvolver a tese do funcionamento da plasticidade cerebral em relação à síndrome de Down.
- Iniciar com tratamentos para estímulo da cognição e evolução motora da criança.
- Estudar como atuar pela inclusão social, atualização de informações para pais e profissionais que trabalham na área.
METODOLOGIA
O trabalho foi desenvolvido com preceitos do estudo exploratório, através de uma pesquisa bibliográfica, onde a pesquisa é constituída a partir de material já existente, através de livros, monografias e artigos científicos.
De acordo com Gil (2006), a maioria das pesquisas acadêmicas realizadas no seu primeiro momento assume a classificação exploratória, pois é difícil em um primeiro momento que o pesquisador tenha uma definição clara do que irá apurar, essa classificação permite aumentar o conhecimento do pesquisador, ao investigar fatos seguindo questões do tipo “o que foi feito”, “como” e “por que”.
Do ponto de vista qualitativo, um fenômeno pode ser mais bem compreendido no contexto em que ocorre e do qual é parte, devendo ser analisado numa perspectiva integrada. Para tanto, o pesquisador vai a campo buscando “captar” o fenômeno em estudo a partir da perspectiva das pessoas nele envolvidas, considerando todos os pontos de vista relevantes. Vários tipos de dados são coletados e analisados para que se entenda a dinâmica do fenômeno. As obras constituem uma fonte não-reativa, as informações nelas contidas permanecem as mesmas após longos períodos de tempo. Podem ser consideradas uma fonte natural de informações à medida que, por terem origem num determinado contexto histórico, econômico e social, retratam e fornecem dados sobre esse mesmo contexto.
O estudo foi realizado no formato de uma revisão de literatura, onde a obtenção de dados ocorreu através de bancos de dados online como: Google Acadêmico, Scielo, PubMed, e através de livros, os termos utilizados na busca foram: Síndrome de Down, cognição, desenvolvimento cognitivo. Como critérios de inclusão foram selecionados artigos disponibilizados completos nos bancos de dados online, entre os anos de 2000 e 2022. Realizou-se uma leitura analítica com o objetivo de ordenar as informações de acordo com sua relevância e uma síntese foi realizada de forma a construir esta monografia.
DESENVOLVIMENTO
A síndrome de Down (SD) é a anomalia cromossômica mais comum; ocorre em 1 a cada 691 bebês, causada pela trissomia do cromossomo 21 e associada a um fenótipo característico em termos de aparência física, bem como a problemas médicos, como distúrbios metabólicos, dismorfismo tecidual, cardiopatia congênita (DAC) e disfunção da tireoide. Normalmente, a superexpressão do cromossomo resulta em uma diminuição do número total de neurônios no sistema nervoso central, um atraso na mielinização e desregulação dos ciclos celulares que levam à superprodução de precursores de proteínas e, posteriormente, causam anormalidades na neurotransmissão. Consequentemente, crianças com SD apresentam comprometimento em domínios cognitivos como concentração, comunicação, memória e desempenho de tarefas em comparação com pares saudáveis. Além disso, as crianças com SD também parecem apresentar um atraso no desenvolvimento motor devido à hipotonia, frouxidão ligamentar, falta de equilíbrio e falta de controle postural que causam dificuldades de adaptação à gravidade e ao ambiente circundante. No desenvolvimento infantil, o funcionamento cognitivo e a psicopatologia estão intimamente interligados. Os anos escolares são um período de neurodesenvolvimento abundante, caracterizado pelo refinamento das habilidades cognitivas enquanto, em algumas crianças, surgem sintomas psiquiátricos. Muitas vezes, uma interrupção em uma área do desenvolvimento é acompanhada por uma deficiência na outra, o que pode refletir um problema subjacente comum de desenvolvimento neurológico. A função cognitiva prejudicada é uma característica central da síndrome de Down. As tentativas de melhorar o funcionamento cognitivo em indivíduos com SD têm enfatizado o aumento da contribuição educacional e de treinamento ou na melhoria da socialização, especialmente na infância e adolescência. De forma consensual os autores expressam que crianças com síndrome de Down tendem a ter maiores dificuldades da linguagem do que seria previsto pela idade mental, enquanto o raciocínio não verbal é uma força relativa. Dentro do domínio da linguagem, as crianças com síndrome de Down mostram uma força relativa na linguagem receptiva em comparação com a linguagem expressiva. A relação entre cognição e psicopatologia é particularmente bem ilustrada por distúrbios do desenvolvimento, como TDAH e TEA, que muitas vezes são caracterizadas por inteligência inferior ou mesmo deficiência intelectual. A criança portadora da síndrome de Down possui o córtex pré-frontal, lobo occipital, hipocampo, tronco cerebral e cerebelo reduzidos (Figuras 1 e 2) em decorrência da triplicada de genes em seu DNA, mais especificamente no cromossomo 21.
Figura 1 – Comparação do encéfalo de recém-natos controle e portadores da Síndrome de Down.
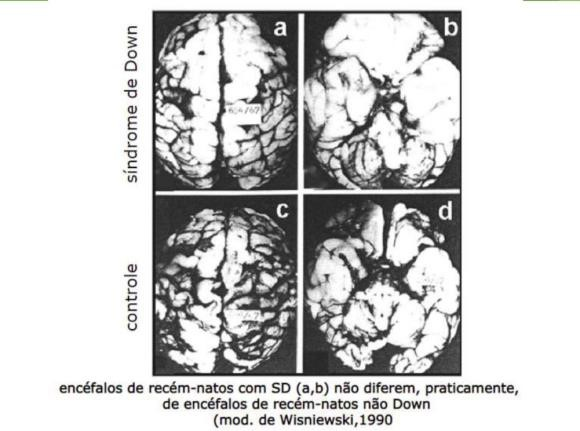
Figura 2 – Comparação do encéfalo de crianças controle e portadores da Síndrome de Down após os 3-5 meses de idade.
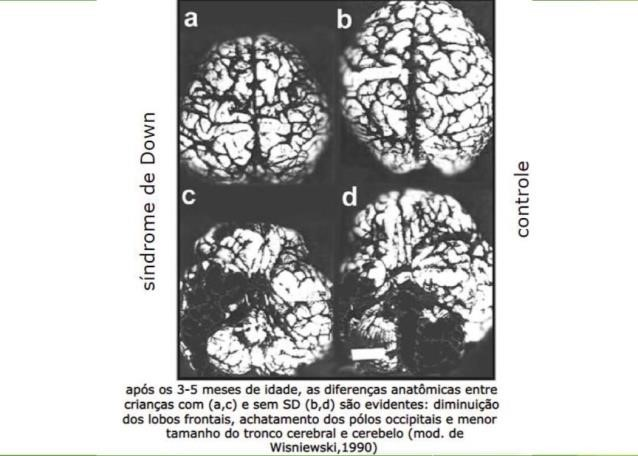
A síndrome de Down é caracterizada por anormalidades na aprendizagem, memória e linguagem que levam a um comprometimento leve a profundo no funcionamento intelectual. Os perfis cognitivos de indivíduos com síndrome de Down são diversos em termos de gravidade da deficiência cognitiva e do tipo de função cognitiva afetada. A morfossintaxe, a memória verbal de curto prazo e a memória explícita de longo prazo geralmente são prejudicadas, e a memória visuoespacial de curto prazo, o aprendizado associativo e a memória implícita de longo prazo geralmente são preservados. Habilidades de linguagem prejudicadas em indivíduos com síndrome de Down podem ter efeitos indiretos no funcionamento de nível superior; portanto, um foco de pesquisa é o desenvolvimento de testes psicométricos e neuropsicológicos que não sejam afetados pelo comprometimento da comunicação. Os efeitos do comprometimento da linguagem na memória verbal de curto prazo e na memória verbal mediada pelo córtex frontal requerem avaliação.
Baroncelli et al. (2011) explicam que o desenvolvimento encefálico da criança diagnosticada com síndrome de Down ocorre de maneira distinta ao desenvolvimento típico. Há a ocorrência da diminuição da maturação do sistema nervoso, vindo a obter pouca definição ocasionando danos espaciais e de função, efeitos estes gerados em consequência da diminuição da quantidade de células diferenciadas. Ademais, os dendritos acabam por atrofiar devido a interrupção em seu desenvolvimento e pode ocorrer a incidência de deficiência mental e motricidade em virtude de alterações sinápticas provenientes da redução da densidade do córtex sensório-motor. Lott e Dierssen (2010) descrevem que o fenótipo cognitivo na síndrome de Down é caracterizado por deficiências na morfossintaxe, memória verbal de curto prazo e memória explícita de longo prazo. No entanto, a memória visuoespacial de curto prazo, a aprendizagem associativa e as funções implícitas da memória de longo prazo são preservadas. As convulsões estão associadas ao declínio cognitivo e parecem causar declínio adicional no funcionamento cognitivo, particularmente em pessoas com síndrome de Down e transtornos comórbidos.
Crianças com síndrome de Down tendem a ter maiores dificuldades da linguagem do que seria previsto pela idade mental, enquanto o raciocínio não verbal é uma força relativa. Dentro do domínio da linguagem, as crianças com síndrome de Down mostram uma força relativa na linguagem receptiva em comparação com a linguagem expressiva. A linguagem tem sido descrita como uma grande área de déficit, com dificuldades particulares decorrentes da linguagem expressiva, gramática e articulação. Embora não exista um teste psicométrico específico para pessoas com síndrome de Down, escalas de inteligência também são utilizadas para fornecer perfis psicológicos. O QI na síndrome de Down está geralmente na faixa de retardo moderado a grave (QI 25-55) (DRUMOND; NASCIMENTO, 2018). Bissoto (2005) complementam que várias características relevantes no que se refere a desenvolvimento cognitivo e linguístico da criança portadora de SD em seus primeiros anos de vida. Por dificuldade na produção da fala, resultam em um vocabulário reduzido, não deixando a criança se expressar ou se comunicar na mesma medida em que compreendem, levando com que as pessoas as subestimem em questão de seu desenvolvimento cognitivo. Além de afetar em outras habilidades. Recursos que utilizem suporte visual para trabalhar as informações; explorando essas habilidades de processamento e de memória visual, poderão beneficiar o ensino de crianças portadoras da Síndrome de Down. Tratar de descobrir todas as oportunidades de oferecer e ensinar para que essas compreendam, buscando sempre melhorias para o desenvolvimento cognitivo e aprendizagem.
Freire, Duarte e Haizin (2012) concluem em sua pesquisa que o funcionamento neuropsicológico da SD é caracterizado pela presença de múltiplas áreas afetadas. No entanto, os conceitos de plasticidade sugerem que podemos criar rotas neuronais e novas estratégias cognitivas que possibilitem a minimização e (ou) superação dos déficits apresentados. A intervenção do tratamento da plasticidade desde cedo apresenta resultados fidedignos. Crianças com a plasticidade neuronal estimulada desde os primeiros anos de vida demonstraram um QI médio de 82, logo, uma diferença qualitativa em relação as crianças com a SD que não praticaram a atividade psicomotora, com QI médio de 62, evidenciando positivamente intervenções desde o princípio da vida de uma criança na Síndrome de Down (CHAKRABARTI et al.2010; CLARK et al. 2006). A tese da plasticidade neuronal envolve o processo no qual uma região cerebral, ao sofrer uma lesão, tem a possibilidade de ser substituída por uma outra porção de células do sistema nervoso central, isto é, neurônios que podem assumir as atividades de células que uma vez foram lesadas e não podem ser reativadas. Visto que a plasticidade dendrítica e axonal são responsáveis pela aprendizagem, cognição e memória, o processo de plasticidade vai depender da idade do indivíduo, tipo de lesão e tamanho do tecido lesionado. Para Nadel (2003) uma diferença imediatamente óbvia é que os cérebros de indivíduos com síndrome de Down são tipicamente menores do que os de controles pareados por idade, pelo menos após os 6 meses de idade. Uma possibilidade que não recebeu atenção suficiente no passado é que essa diferença é consequência do fato de os indivíduos com síndrome de Down terem corpos menores. Ou seja, as diferenças no tamanho do cérebro podem ser uma questão de alometria. Essa possibilidade, somada ao fato de que não existe uma relação clara entre o tamanho do cérebro e a “inteligência” em qualquer caso, sugere que o retardo mental observado na síndrome de Down resulta de algo além de diferenças grosseiras no tamanho do cérebro. O remanejamento do sistema nervoso central é a alteração de estruturas corticais existentes, funções cognitivas, motoras, processos fisiológicos e novo mapeamento cerebral através do estímulo. A super estimulação em camundongos afirma que a instrução e retirada de sinapses é possível, havendo uma rotatividade sináptica por estímulo, gerando um novo mapeamento neurológico (COSTA,2019).
O ambiente no qual a criança está inserida possui grande importância em seu desenvolvimento cognitivo, muito mais se este a proporcione condições para aprendizagem possibilitando que sejam realizados estímulos que contribuam para sua evolução intelectual, além de que é de suma importância a participação de seus familiares na execução de atividades e brincadeiras em locais em que sejam favoráveis para a criança para que, além do seu desenvolvimento mental, seja possível a socialização e a aprendizagem de modo que contribua para sua capacidade motora, verbal e cognitiva (BRAULT et al.2021; PICKER; WALSH, 2013).
Mancini et al. (2003) demonstraram em seu estudo que o desempenho funcional de crianças com SD é inferior ao de crianças normais. Entretanto, as interações entre patologia e grupo etário revelaram que este desempenho inferior não se mantém constante no contínuo do desenvolvimento. As crianças do grupo SD apresentaram um repertório de habilidades funcionais inferior e mostraram-se mais dependentes da ajuda fornecida pelo cuidador, quando comparadas com crianças com desenvolvimento normal, nas três áreas de função.
Os problemas de aprendizagem e memória que começam a surgir no final da infância tornam-se consideravelmente mais perceptíveis à medida que a criança cresce até a infância e a adolescência. Embora grande parte do nosso conhecimento para esse período venha do aprendizado da linguagem, há informações disponíveis sobre outros tipos de aprendizado e memória. Um ponto importante a ser enfatizado a partir desses dados de aprendizagem de línguas tem menos a ver com a incapacidade das crianças com síndrome de Down de adquirir palavras, ou construções linguísticas, ou outro material não verbal, e mais a ver com sua incapacidade de ‘estabilizar’ as informações que eles conseguem adquirir. A relação entre os efeitos genômicos da trissomia que causa a síndrome de Down e os mecanismos patogenéticos subjacentes aos déficits cognitivos não são totalmente compreendidos, em parte devido à ampla variabilidade dos efeitos fenotípicos do distúrbio. Essa variabilidade pode ser causada por variação alélica, desequilíbrio genômico ou fatores epigenéticos ou ambientais, e, portanto, alterações estruturais homólogas no cérebro não afetam todos os indivíduos com síndrome de Down da mesma forma caminho.
As famílias desempenham um papel fundamental na intervenção precoce e no desenvolvimento futuro de seus filhos. Sabemos que a intervenção precoce é mais eficaz se a família for um agente ativo na sua implementação. Ao considerar as habilidades de aprendizagem e memória de indivíduos com síndrome de Down, duas grandes preocupações devem ser levadas em conta: primeiro, aprendizagem e memória devem ser consideradas como envolvendo vários sistemas separados; e segundo, atenção deve ser dada às habilidades dos indivíduos em vários estágios da vida.
Silva e Kleinhans (2006) ainda relatam que os conceitos de plasticidade sugerem que podemos substituir uma função exercida por uma área lesada do cérebro por outra não lesada ou menos lesada. Como pudemos perceber, na SD, muitas áreas, se não todas, podem apresentar algum tipo de alteração. Mas as pesquisas também mostraram que há diferenças entre um sujeito e outro, tanto na intensidade como na área lesada, o que nos leva a concluir que se um trabalho de estimulação dos processos cognitivos for realizado de maneira adequada, nos primeiros anos de vida, poderá promover significativas modificações qualitativas no desenvolvimento.
A aprendizagem exige respostas que podem ser motoras, verbais ou gráficas. A resposta manifestada pela criança com SD será pobre devido às limitações que apresenta. Contudo, a possibilidade de ampliar e determinar certa resposta estará condicionada ao apoio do meio. Quanto mais se oferecer um ambiente solicitador, que promova autonomia e diferentes possibilidades de descobertas de seu potencial, melhor será o seu desenvolvimento. Reconhecendo as características do fenótipo de pessoas com SD, deveríamos concentrar as atividades nas áreas em que há maior potencial. Assim, na medida em que o sujeito percebe que pode realizar determinadas tarefas com êxito, haverá satisfação e maior motivação para enfrentar aquelas que ele tem maior dificuldade, contribuindo para que, dessa forma, seu desenvolvimento físico e mental vá avançando passo a passo. A educação requer paciência, dedicação e firmeza, sobretudo carinho e amor de pais e profissionais. Todos têm habilidades e dificuldades, apenas precisamos conhecê-las e aprender a lidar com elas.
A síndrome de Down (SD) está associada a uma variedade de sintomas, como retardo mental incapacitante e neuro degeneração (ou seja, doença de Alzheimer), que impedem os pacientes de levar uma vida totalmente independente. Esses fenótipos são uma consequência direta da superexpressão dos genes do cromossomo 21, que estão presentes em duplicata devido à não disjunção do cromossomo 21. Dados acumulados sugerem que o produto do gene do cromossomo 21, tirosina-(Y) fosforilação de dupla especificidade regula a quinase 1A (Dyrk1A), participantes dos mecanismos patogênicos subjacentes aos sintomas mentais e outros sintomas físicos da SD (STRINGER, 2017).
Segundo Brault (2021) a síndrome de Down é devido a uma trissomia completa Hsa21 ou uma trissomia parcial que inclui a região crítica 21q22.3. 95% dos casos são devidos a trissomia completa ou regular; cerca de 3% é devido ao mosaicismo, distúrbio em que os pacientes apresentam células normais e células com Hsa21 extra juntas; menos de 2% é causado por uma translocação desequilibrada; ou seja, um cariótipo com 46 cromossomos, mas um deles, geralmente o cromossomo 14, contém material cromossômico extra de Hsa21. A Organização Mundial da Saúde estima uma prevalência mundial de 1 em cada 1.000 nascidos vivos; no entanto, esses números variam, refletindo que a prevalência depende de variantes socioculturais, como acesso ao diagnóstico prénatal e interrupção legal da gravidez.
O diagnóstico é clínico e confirmado por citogenética. O padrão de características físicas observáveis é altamente sugestivo, assim como as alterações sistêmicas. No entanto, nem todas as alterações estão presentes em todos os indivíduos afetados. Em recém-nascidos o diagnóstico pode ser difícil; no entanto, dez características são altamente prevalentes. Hall, em 1966, analisou 48 recém nascidos afetados e descobriu que 100% tinham 4 ou mais características e 89% tinham 6 ou mais. Desde então, essas características têm sido utilizadas para avaliar todos os recém-nascidos vivos, conhecidos como critérios de Hall (PARK, 2013).
Existem vários genes na região crítica para a síndrome de Down. O gene DYRK1A (21q22.13) que é expresso no sistema nervoso em desenvolvimento de adultos, tem como função a inibição da proliferação celular e promoção da diferenciação neuronal prematura. Estudos em camundongos que super expressam Dyrk1a mostraram graves deficiências de aprendizado, bem como defeitos de memória espacial (NEUMANN, 2018). Da mesma forma, o gene SIM2 (21q22.13), ortólogo ao gene de Drosophila é um fator de transcrição e o principal regulador do desenvolvimento; também é expresso no cérebro humano em desenvolvimento e em camundongos transgênicos. Eles mostraram dificuldades de aprendizado leves e problemas de memória. A molécula de adesão da síndrome de Down (DSCAM, 21q22.2) é expressa em dendritos neuronais e contribui para a plasticidade sináptica; no entanto, inibe a ramificação dendrítica quando super expressa em neurônios do hipocampo in vitro e no camundongo com três cópias de Dscam (STENSEN, 2021).
Existem também genes fora da região crítica da síndrome de Down que têm sido associados ao fenótipo neurológico de pacientes com síndrome de Down. Synaptojanin1 (SYNJ1, 21q22.2) é uma proteína formadora de vesículas em sinapses neuronais que desempenha um papel importante na neurotransmissão por desfosforilação de fosfatidilinositol bifosfato alterado em um modelo de camundongo com síndrome de Down que tinha problemas de aprendizado e memória, que foram normalizados pela redução do gene Synj1 de três para dois (ATAS-OZCAN, 2021). Por fim, a análise das trissomias segmentares confirma o importante papel da proteína precursora de amilóide (APP, 21q21.3) uma vez que os inibidores dos metabólitos dessa proteína, em modelo de camundongo, melhoraram seu aprendizado e memória, sugerindo que a dose tripla do gene APP poderia ser a causa do fenótipo neurológico em pacientes com síndrome de Down (BHANSALI,2021).
Neonatos com síndrome de Down comumente apresentam hipotonia e a maioria das alterações motoras. Achados em humanos e modelos de camundongos mostraram um número reduzido de neurônios granulares no cerebelo (THOMAS, 2021). Essa neurogênese cerebelar reduzida pode ser devido a um defeito na sinalização sonic hedgehog (SHH) em neurônios precursores, causado por níveis elevados de APP. 24 Outro gene já citado é o DYRK1A, que também é proposto como gene candidato para déficit motor nesses pacientes (BHANSALI, 2021).
Várias linhas de evidência sugerem um papel chave de DYRK1A no controle do crescimento celular, neurogênese e maturação neuronal. A proteína p53 é um dos alvos de DYRK1A. Em particular, DYRK1A demonstrou fosforilar p53 levando à regulação positiva de genes alvo p53, incluindo p21 CIP1 (também conhecido como inibidor 1 de quinase dependente de ciclina ou proteína que interage com CDK), uma proteína envolvida na regulação do ciclo celular. A regulação positiva de p21 CIP1 prejudica a transição de fase G1/G0-para-S, inibindo a proliferação de células neuroprogenitoras (NPC) (STRINGER, 2017). Consistente com isso, níveis aumentados de p21CIP1 foram encontrados em cérebros de camundongos transgênicos Dyrk1a e de fetos com SD (STENSEN, 2021). Dyrk1a é ubiquamente expresso em diferentes populações de células neuronais do cérebro, mas com diferenças regionais: o nível de proteína é maior no bulbo olfatório, córtex cerebelar, estruturas corticais e camadas de células granulares e piramidais do hipocampo. Foi verificado a expressão de DYRK1A em neurônios glutamatérgicos adultos, por localização co-imunohistoquímica com um anticorpo contra CAMK2A e DYRK1A. No camundongo adulto do tipo selvagem, ambas as proteínas foram encontradas em neurônios piramidais e granulares do hipocampo e giro denteado e em neurônios do córtex (NEUMANN, 2018).
Atas-Ozcan (2021) diz que a contribuição de DYRK1A para a regulação da dinâmica da actina foi observada em Levedura, com seu homólogo de Schizosaccharomyces pombe interagindo com a proteína ativadora de GTPase RGA4 e contribuindo para a localização adequada de RGA4 e do regulador Factina CDC42 GTPase para locais corticais da célula onde as estruturas F-actina são localizadas para gerar polaridade celular e com seu homólogo Mnb identificado em uma tela de RNAi de quinoma com base na morfologia celular como um modulador da protrusão baseada em actina. O controle da montagem de filamentos de actina por DYRK1A durante o desenvolvimento de dendritos neuronais e espinhas dendríticas ocorre através da fosforilação múltipla de treonina da proteína N-WASP dentro de seu domínio de ligação a GTPase, levando à redução da montagem de filamentos de actina mediada por Arp2/3.
Thomas (2021), relata que Dyrk1A também contribui para a regulação da dinâmica da actina e sinaptogênese. O homólogo de levedura da Dyrk quinase, Pom1, interage com a proteína ativadora de Rga4 GTPase e regula a localização de Rga4, que está envolvida na localização de Cdc42 GTPase em levedura. A tela de interferência de RNA do quinoma de Drosophila com base na morfologia celular identificou o minicérebro (homólogo de Drosophila de Dyrk1A) como um regulador da organização da actina. O knockdown específico do minicérebro causou um aumento na actina periférica e no número de saliências na linhagem celular derivada do SNC. Enquanto o knockdown específico de Dyrk1A em células COS-7 promoveu a formação de filopódios (projeções citoplasmáticas), a superexpressão de Dyrk1A causou uma redução na formação de espinhas dendríticas de neurônios hipocampais cultivados. Em forte correlação, os neurônios corticais de camundongos transgênicos Dyrk1A exibiram uma redução na densidade da coluna, formação de sinapses e comprimento dos filopódios dendríticos, bem como alteração na morfologia da coluna. A associação de Dyrk1A com filamentos de actina pode ser ainda apoiada por ensaios de coimunoprecipitação com tecidos DS.
A interação intramolecular de NWASP é um regulador primário da atividade de N-WASP. Assim, o domínio VCA de N-WASP pode ativar Arp2/3 a jusante, enquanto sua ação é auto-inibida por sua interação com a região GBD. Dados de coimunoprecipitação e experimentos de ligação in vitro usando fragmentos N-WASP VCA e BGBD, e testando o efeito de seu mutante de substituição de glutamato fosfomimético revelam que o resíduo Thr259 é o local de direcionamento mais eficaz de Dyrk1A. Este site regula especificamente a interação intramolecular de N-WASP (NEUMANN, 2018).
O domínio de ligação à GTPase de N-WASP interage com a pequena GTPase Cdc42 a montante. A ligação ao Cdc42 altera a estrutura da proteína de NWASP; isso ativa a sinalização mediada por Arp2/3 a jusante, que subsequentemente leva à montagem de filamentos de actina. Primeiro se a fosforilação de Dyrk1A afeta a afinidade de ligação de NWASP para Cdc42. As células HEK293 foram cotransfectadas com plasmídeos que codificam Cdc42 constitutivamente ativo (V12) fundido a GST e um mutante NWASP marcado com FLAG, que tem uma substituição de glutamato no(s) resíduo(s) Thr alvo, que imita o efeito de Dyrk1A mediados fosforilações em N-WASP. A análise de imunotransferência dos precipitados suspensos com anticorpo anti-FLAG mostrou que três proteínas N-WASP com uma única fosforilação (T196E, T202E e T259E) não se ligam diferencialmente a Cdc42, em comparação com WT N-WASP. Para examinar os efeitos cooperativos das múltiplas fosforilações, realizamos ensaios de co-imunoprecipitação adicionais usando os mutantes com substituições duplas nos resíduos T196E e T202E próximos ou substituições triplas em T196E, T202E, T259E (3TE). A ligação desses dois mutantes com substituições duplas ou triplas de glutamato a Cdc42 não foi afetada. Para excluir a possibilidade de efeitos de ligação por proteínas endógenas não identificadas, testamos adicionalmente a ligação in vitro de WT N-WASP ou mutante 3TE purificado por bactérias a Cdc42 (V12) purificado de lisados de células HEK293. A análise de imunotransferência dos precipitados pull-down com anticorpo anti-His confirmou que a ligação de N-WASP a Cdc42 não é afetada por Dyrk1A (SANTANA; CAVALCANTE, 2018).
A trissomia 21 causa um lastro efeito no desenvolvimento do cérebro, ação de distúrbios no neurodesenvolvimento, como uma disfunção sináptica, causando um déficit de aprendizagem e memória. Várias funções prejudicadas, em alguns neurotransmissores como; Gaba e alterações nos receptores de glutamato, também incluem variações nas proteínas sinápticas. Causando prejuízo no mecanismo de sinapse. Por isso a importância da plasticidade, que podem trazer benefícios de mudanças na estrutura física da sinapse (MARTINEZ, 2019). Ao longo da última década, avanços foram feitos na compreensão da genética do desempenho cognitivo e da disfunção cognitiva na deficiência intelectual sindrômica e não sindrômica. Na deficiência intelectual sindrômica, foram identificados mecanismos moleculares específicos que podem contribuir para a cognição prejudicada e levantam a possibilidade de que tratamentos direcionados a esses mecanismos possam ser desenvolvidos para melhorar o desempenho cognitivo em indivíduos com síndrome de Down (SANTANA; CAVALCANTE, 2018).
Crianças com síndrome de Down têm um fenótipo cognitivo distinto caracterizado por um padrão particular de déficits, bem como um padrão de forças relativas, quando comparadas às crianças desenvolvimento típico e crianças com outros tipos de comprometimento cognitivo. De um modo geral, as crianças com síndrome de Down parecem ter um perfil particular de memória e habilidades de processamento de informações, incluindo capacidade reduzida de memória de trabalho, memória implícita intacta, mas memória de longo prazo reduzida para informações explícitas e processamento verbal mais pobre e habilidades de memória auditiva de curto prazo em relação à idade mental não verbal e forças relativas no processamento visuoespacial e memória não verbal (VOIVODIC; STORER, 2002).
Além disso, avanços teóricos e uma abordagem desenvolvimentista da deficiência intelectual levaram à caracterização de um fenótipo cognitivo para a síndrome de Down. Tem sido sugerido que compreender a trajetória do desenvolvimento cognitivo em crianças com síndrome de Down pode levar ao desenho de intervenções mais efetivas (SANTANA; CAVALCANTE, 2018).
As alterações apresentadas por crianças portadoras de SD podem se manifestar funcionalmente interferindo na capacidade destas crianças de desempenhar de forma independente diversas atividades e tarefas da rotina diária. Embora a literatura disponibilize evidências sobre as limitações consequentes desta condição genética em termos das funções de órgãos e sistemas que compõem a estrutura do corpo destas crianças (capacidades motoras e cognitivas), informações sobre o impacto destas limitações internas no desempenho de atividades diárias deste grupo são menos frequentes. Entretanto, esse tipo de informação funcional é extremamente relevante para profissionais da área da saúde uma vez que as expectativas dos pais de crianças portadoras de SD estão mais relacionadas à informação funcional do que a informação sobre sintomatologia e componentes específicos de desempenho. A escassez de evidências sobre o desempenho funcional deste grupo clínico limita os profissionais que lidam com estas crianças a predizer desfechos e expectativas possíveis de serem alcançadas (MANCINI et al.2003).
CONSIDERAÇÕES FINAIS
O presente estudo atingiu o objetivo ao concluir que indivíduos com síndrome de Down (SD) geralmente possuem perfis neurocognitivos e neurocomportamentais únicos que surgem em períodos específicos de desenvolvimento. Esses perfis são distintos em relação a outros com deficiência intelectual (DI) semelhante e refletem achados neuroanatômicos subjacentes, fornecendo suporte para um perfil fenotípico distinto. Na primeira infância, surgem leves desvios das trajetórias de desenvolvimento neurotípico. Na idade escolar, os atrasos tornam-se pronunciados. As habilidades não verbais permanecem na trajetória para a idade mental, enquanto os déficits verbais emergem e persistem. A aprendizagem não-verbal e a memória são pontos fortes em relação às habilidades verbais. A linguagem expressiva é atrasada em relação à compreensão. Aspectos das habilidades linguísticas continuam a se desenvolver ao longo da adolescência, embora as habilidades linguísticas permaneçam comprometidas na idade adulta. Déficits de atenção/funções executivas estão presentes na infância e tornam-se mais pronunciados com a idade. Os traços característicos associados à SD (alegria, natureza social) são ativos de personalidade.
Indivíduos com SD possuem muitos pontos fortes e fracos únicos que devem ser apreciados à medida que se desenvolvem ao longo da vida. A conscientização desse perfil por profissionais e cuidadores pode promover a detecção precoce e apoiar o desenvolvimento cognitivo e comportamental desse crianças portadoras da síndrome.
REFERÊNCIAS BIBLIOGRÁFICAS
- COELHO, Charlotte. A síndrome de Down. Psicologia. pt, p. 1-14, 2016. Disponível em: https://www.psicologia.pt/artigos/textos/A0963.pdf. Acesso em: 07 de junho de 2022.
- BISSOTO, Maria Luísa. O desenvolvimento Cognitivo e o processo de aprendizagem do portador de síndrome de Down: revendo concepções e perspectivas educacionais. Ciências & Cognição, v. 4, p. 80-88, 2005. Disponível em: http://pepsic.bvsalud.org/scielo.php?script=sci_arttext&pid=S1806- 58212005000100009
- DRUMOND, Adriana; NASCIMENTO, Rodolfo Francisco do. Aspectos da linguagem na criança com Síndrome de Down: influências no processo da fala e alfabetização. Mal-Estar e Sociedade, v. 8, n. 2, 2018.
- CHAKRABARTI, Lina et al. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nature neuroscience, v. 13, n. 8, p. 927-934, 2010.
- CLARK, Sarah et al. Fluoxetine rescues deficient neurogenesis in hippocampus of the Ts65Dn mouse model for Down syndrome. Experimental neurology, v. 200, n.1, p. 256-261, 2006.
- COSTA, Daniela Ferreira da. Síndrome de Down: Estratégias Terapêuticas para melhoria dos défices cognitivos. 2019. Tese de Doutorado. Universidade de Coimbra.
- BRAULT, Véronique et al. Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down syndrome. PLoS genetics, v. 17, n. 9, p. e1009777, 2021.
- PICKER, Jonathan D.; WALSH, Christopher A. New innovations: therapeutic opportunities for intellectual disabilities. Annals of neurology, v. 74, n. 3, p.382390, 2013.
- PARK, Joongkyu; CHUNG, Kwang Chul. New perspectives of Dyrk1A role in neurogenesis and neuropathologic features of Down syndrome. Experimental neurobiology, v. 22, n. 4, p. 244, 2013.
- STENSEN, Wenche et al. Novel DYRK1A Inhibitor Rescues Learning and Memory Deficits in a Mouse Model of Down Syndrome. Pharmaceuticals, v. 14, n. 11, p.1170, 2021.
- ATAS-OZCAN, Helin et al. Dyrk1a from Gene Function in Development and Physiology to Dosage Correction across Life Span in Down Syndrome. Genes, v. 12, n. 11, p.1833, 2021.
- BHANSALI, Rahul S. et al. DYRK1A regulates B cell acute lymphoblastic leukemia through phosphorylation of FOXO1 and STAT3. The Journal of clinical investigation, v. 131, n. 1, 2021.
- THOMAS, Jared R. et al. Skeletal Deficits in Male and Female down Syndrome Model Mice Arise Independent of Normalized Dyrk1a Expression in Osteoblasts. Genes, v. 12, n. 11, p. 1729, 2021
- STRINGER, Megan; GOODLETT, Charles R.; ROPER, Randall J. Targeting trisomic treatments: optimizing Dyrk1a inhibition to improve Down syndrome deficits. Molecular genetics & genomic medicine, v. 5, n. 5, p. 451-465, 2017.
- NEUMANN, Fernanda et al. DYRK1A inhibition and cognitive rescue in a Down syndrome mouse model are induced by new fluoro-DANDY derivatives. Scientific reports, v. 8, n. 1, p. 1-12, 2018.
- SANTANA, Nayara Xavier; CAVALCANTE, Jordano. Conceito neuroevolutivo em pacientes com síndrome de down: revisão integrativa. SALUSVITA, Bauru, v. 37, n. 4, p. 1009-1018, 2018. Disponível em: https://secure.unisagrado.edu.br/static/biblioteca/salusvita/salusvita_v37_n4_201 8/sa lusvita_v37_n4_2018_art_15.pdf. Acesso em: 07 de junho de 2022.
- CUÉ, Carmen Martinez; Dierssen, Mara. Plasticity as a therapeutic target for improving cognition and behavior in Down syndrome. Chapter 9, 2019.
- VOIVODIC, Maria Antonieta; STORER, Márcia Regina de Souza. O desenvolvimento cognitivo das crianças com síndrome de Down à luz das relações familiares. Psicologia: teoria e prática, v. 4, n. 2, p. 31-40, 2002. Disponível em: http://pepsic.bvsalud.org/scielo.php?script=sci_arttext&pid=S151636 872002000200004. Acesso em: 07 de junho de 2022.
- MANCINI, Marisa Cotta et al. Comparação do desempenho funcional de crianças portadoras de síndrome de Down e crianças com desenvolvimento normal aos 2 e 5 anos de idade. Arquivos de Neuro-Psiquiatria, v. 61, p. 409-415, 2003.
- ANHÃO, Patrícia Páfaro Gomes; PFEIFER, Luzia Iara; SANTOS, Jair Lício dos. Interação social de crianças com Síndrome de Down na educação infantil. Revista Brasileira de Educação Especial, v. 16, n. 1, p. 31-46, 2010. Disponível em: https://www.scielo.br/j/rbee/a/Zp9WPjxhLMKDnBX3ZgWN9Tk/?format=pdf&lang=pt. Acesso em: 07 de junho de 2022.
- BARONCELLI, Laura et al. Brain plasticity and disease: a matter of inhibition. Neural plasticity, v. 2011, 2011
- COSTA, Alan Ricardo; DA SILVA, Peterson Luiz Oliveira; JACÓBSEN, Rafael Tatsch. Plasticidade cerebral: conceito (s), contribuições ao avanço científico e estudos brasileiros na área de Letras. Entrepalavras, v. 9, n. 3, p. 457-476, 2019.
- FREIRE, Rosália Carmen; DUARTE, Nietsnie; HAZIN, Izabel. Fenótipo neuropsicológico de crianças com síndrome de Down. Psicologia em Revista, v.18, n. 3, p. 354-372, 2012.
- HYDE, Lynn A.; FRISONE, Deborah F.; CRNIC, Linda S. Ts65Dn mice, a model for Down syndrome, have deficits in context discrimination learning suggesting impaired hippocampal function. Behavioural brain research, v. 118, n. 1, p. 53-60, 2001.
- LOTT, Ira T; DIERSSEN, Mara. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neural, v.9, p623-33, 2010. Disponível em: https://www.thelancet.com/journals/laneur/article/PIIS1474442210701125/fulltext#:~:t ext=The%20four%20ages%20of%20Down%20syndrome.&text=The%20disorder%2 0is%20caused%20by,neurological%20complications%20of%20systemic%20disorder
- MAIA, Joviane Marcondelli Dias; WILLIAMS, Lucia Cavalcanti de Albuquerque. Fatores de risco e fatores de proteção ao desenvolvimento infantil: uma revisão da área. Temas em psicologia, v. 13, n. 2, p. 91-103, 2005. Disponível em:https://www.redalyc.org/pdf/5137/513751425002.pdf. Acesso em: 07 de junho de 2022.
- NADEL, Lynn. Down’s syndrome: a genetic disorder in biobehavioral perspective.Genes, Brain and Behavior, v. 2, n. 3, p. 156-166, 2003.
- PENNINGTON, Bruce F. et al. The neuropsychology of Down syndrome: evidence for hippocampal dysfunction. Child development, v. 74, n. 1, p. 75-93, 2003.
- SALE, Alessandro; BERARDI, Nicoletta; MAFFEI, Lamberto. Enrich the environment to empower the brain, 2009.
- SILVA, Nara Liana Pereira; DESSEN, Maria Auxiliadora. Síndrome de Down: etiologia, caracterização e impacto na família. Interação em psicologia, v. 6, n. 2, 2002. Disponível em: https://revistas.ufpr.br/psicologia/article/view/3304. Acesso em: 07 de junho de 2022.
- SILVA, Maria de Fátima Minetto Caldeira; KLEINHANS, Andréia Cristina dos Santos. Processos cognitivos e plasticidade cerebral na Síndrome de Down. Revista Brasileira de educação especial, v. 12, p. 123-138, 2006.
- SOUCHET, Benoit et al. Prenatal treatment with EGCG enriched green tea extract rescues GAD67 related developmental and cognitive defects in Down syndrome mouse models. Scientific reports, v. 9, n. 1, p. 1-13, 2019.
- TRINDADE, André Soares; NASCIMENTO, Marcos Antonio do. Avaliação do desenvolvimento motor em crianças com síndrome de down. Revista Brasileira de Educação Especial, v. 22, p. 577-588, 2016.
- TORRES, Andreia Araujo Lima. Aspectos nutricionais associados ao envelhecimento de indivíduos com síndrome de Down: uma revisão integrativa. Revista Brasileira de Ciências do Envelhecimento Humano, v. 13, n. 3, 2016.
- THOMAS, Jared R. et al. Skeletal Deficits in Male and Female down Syndrome Model Mice Arise Independent of Normalized Dyrk1a Expression in Osteoblasts. Genes, v. 12, n. 11, p. 1729, 2021. Biomedicina – 8º semestre – 2022/2 24
Filiação:
Discente da Universidade Anhembi Morumbi, São Paulo / SP, Brasil
Docente da Universidade Anhembi Morumbi, São Paulo / SP, Brasil