EVALUATION OF PHYSICOCHEMICAL QUALITY CONTROL ASSAYS OF ACETYLSALICYLIC ACID MEDICATION: A COMPARATIVE ANALYSIS BETWEEN THE RESULTS OF THE REFERENCE MEDICATION AND GENERICS
REGISTRO DOI: 10.5281/zenodo.11522666
Ana Patrícia Silva dos Santos1,
Matheus Gomes Oliveira2,
Darlan Ferreira de Souza3,
RESUMO
É fundamental garantir a qualidade e segurança dos medicamentos, o controle de qualidade de uma indústria farmacêutica atua na detecção de possíveis desvios e erros que possam comprometer os critérios de segurança para o paciente, seguindo as regulamentações nacionais e internacionais, como as boas práticas de fabricação. A Anvisa estabelece diretrizes para o controle de qualidade na indústria farmacêutica, incluindo análises físico-químicas e microbiológicas. No caso específico do ácido acetilsalicílico, amplamente utilizado como analgésico e antipirético, a análise de dissolução é essencial para verificar sua biodisponibilidade e eficácia terapêutica, por exemplo. O objetivo deste estudo foi de avaliar e comparar a qualidade em medicamentos contendo ácido acetilsalicílico, por meio de resultados dos testes físicos-químicos. Foi realizado aquisição de comprimidos de AAS do medicamento referência (Laboratório 1) e dois genéricos de laboratórios distintos (Laboratório 2 e Laboratórios 3) para comparação dos seguintes parâmetros: peso médio, friabilidade, dureza, desintegração, doseamento e dissolução seguindo os parâmetros da Farmacopeia Brasileira 6ª edição. O laboratório 1, 2 e 3 cumpriram o teste de determinação de peso onde foram apresentados as médias e o desvio padrão e relativo e não se ultrapassou de 1,5%. Na friabilidade foi calculada a perda de peso (%) e ambos passaram na avaliação, assim como, a dureza que possui caráter informativo, na qual, foi analisada em relação à média sendo os valores obtidos aceitáveis. Seguindo o parâmetro exigido para o tempo de desintegração que é de 5 min, ambos estão aprovados. Para os testes de doseamento, obteve os seguintes resultados respectivamente, 98,50%, 96,72% e 95,04%, e estão aprovados segundo a Farmacopeia Brasileira. Por fim, as avaliações de dissolução feitas com os laboratórios também obtiveram resultados positivos, sendo eles acima de 80% como preconizado. Destacamos a relevância desses testes na garantia da qualidade e eficácia terapêutica dos medicamentos. Os resultados obtidos confirmam que as amostras dos laboratórios 1, 2 e 3 estão adequadas para o consumo, tanto em termos de inserção quanto de distribuição seguindo especificações previamente preconizadas pela Farmacopeia. É importante ressaltar a necessidade desses testes para garantir a qualidade, segurança e efetividade terapêutica deste medicamento.
Palavras-chave: Ácido Acetilsalicílico. Anvisa. Controle de Qualidade. Medicamento.
ABSTRACT
It is essential to ensure the quality and safety of medications. The quality control of a pharmaceutical industry plays a crucial role in detecting possible deviations and errors that may compromise safety criteria for patients, following national and international regulations such as good manufacturing practices. Anvisa establishes guidelines for quality control in the pharmaceutical industry, including physicochemical and microbiological analyses. In the specific case of acetylsalicylic acid, widely used as an analgesic and antipyretic, dissolution analysis is essential to verify its bioavailability and therapeutic efficacy. The objective of this study was to evaluate and compare the quality of medications containing acetylsalicylic acid through physicochemical test results. Tablets of acetylsalicylic acid were acquired from the reference medication (Laboratory 1) and two different generic products from distinct laboratories (Laboratory 2 and Laboratory 3) for comparison of the following parameters: average weight, friability, hardness, disintegration, assay, and dissolution following the parameters of the Brazilian Pharmacopoeia 6th edition. Laboratories 1, 2, and 3 passed the weight determination test, where the means and standard deviations were presented, without exceeding 1.5%. Friability was calculated as a percentage of weight loss, and both passed the evaluation, as did the hardness, which had an informative character, with the obtained values being acceptable. Meeting the required parameter of a disintegration time of 5 minutes, both products are approved. For the assay tests, the following results were obtained, respectively: 98.50%, 96.72%, and 95.04%, all approved according to the Brazilian Pharmacopoeia. Finally, the dissolution evaluations conducted with the laboratories also yielded positive results, with values above 80% as recommended. We emphasize the relevance of these tests in ensuring the quality and therapeutic efficacy of medications. The obtained results confirm that the samples from Laboratories 1, 2, and 3 are suitable for consumption, both in terms of production and distribution, following previously established specifications by the Pharmacopoeia. It is important to highlight the necessity of these tests to guarantee the quality, safety, and therapeutic effectiveness of this medication.
Key-words: Acetylsalicylic acid. Anvisa. Quality Control. Medicine.
INTRODUÇÃO
O controle de qualidade na indústria farmacêutica é fundamental por garantir a segurança e eficácia dos medicamentos produzidos, além de atender às normas regulatórias estabelecidas pelos órgãos competentes. Segundo Chen et al. (2020), o controle de qualidade engloba diversas etapas, desde o desenvolvimento do produto até a sua comercialização, incluindo a escolha adequada das matérias-primas, o monitoramento da produção, a realização de testes. Além disso, Maia et al. (2016) destacam a importância deste setor para a detecção precoce de desvios e falhas no processo produtivo, prevenção de erros que possam comprometer a qualidade do produto final e na melhoria contínua dos processos. Isso é fundamental não somente para evitar prejuízos financeiros, mas principalmente para proteger a saúde e bem-estar dos pacientes.
Ainda de acordo com Silva et al. (2021), o controle de qualidade na indústria farmacêutica deve estar alinhado com as regulamentações nacionais e internacionais, como as boas práticas de fabricação (BPF) e as diretrizes da Agência Nacional de Vigilância Sanitária (ANVISA). Em resumo, o controle de qualidade na indústria farmacêutica é essencial por garantir a segurança e eficácia dos medicamentos produzidos, atender às normas regulatórias estabelecidas e proteger a saúde dos pacientes. Neste sentido, há forte papel no monitoramento dos requisitos preconizados pelos órgãos reguladores, estas exigências são garantidas por uma série de análises.
A Resolução da Diretoria Colegiada – RDC nº 658/2022 da Anvisa, dispõe sobre as boas práticas de fabricação de medicamentos. Em relação às análises físico-químicas, a RDC estabelece que os materiais usados na produção devem ser submetidos à identificação, avaliação e controle, por meio de ensaios físico-químicos específicos, antes de serem utilizados no processo produtivo. Ademais, a RDC prevê que as amostras dos medicamentos produzidos devem ser submetidas a testes físico-químicos, microbiológicos e de estabilidade, para garantir a qualidade e eficácia do produto (NACIONAL, 2022).
A aplicação dessas diretrizes no controle de qualidade do ácido acetilsalicílico, fármaco este que é amplamente utilizado no tratamento de dores e febres e possui fácil acesso pela população. Além disso, este medicamento é isento de prescrição médica, cuja qualidade e segurança são garantidas por uma série de análises físico-químicas, como, por exemplo, a técnica de dissolução, uma pesquisa publicada em 2020 no Journal of Drug Delivery Science and Technology destaca a importância da análise de dissolução do ácido acetilsalicílico para verificar sua biodisponibilidade e eficácia terapêutica (RODRIGUES, T., et al., 2020).
METODOLOGIA
Os ensaios apresentados neste estudo foram realizados no laboratório de química analítica do Centro Universitário Augusto Motta entre o período de abril a junho, seguindo as especificações da Farmacopeia Brasileira 6ª edição. Os ensaios foram feitos em comprimidos de AAS 100 mg dos diferentes tipos, sendo eles: medicamento referência denominado de laboratório 1, um medicamento genérico chamado de laboratório 2 e um segundo genérico sendo ele de outro laboratório, nomeado de laboratório 3. Dessa forma, os testes realizados foram de: Determinação de Peso Médio, Friabilidade, Dureza, Doseamento, Desintegração e Dissolução. Todos os respectivos ensaios seguiram especificações farmacopeicas.
Determinação de peso
Foi utilizada uma balança analítica para realizar a pesagem individual de 20 comprimidos de AAS de 100mg, com o objetivo de determinar o peso médio. Neste experimento, é permitido que no máximo duas unidades estejam fora do intervalo de variação de ± 7,5%, e nenhuma unidade pode ter um peso acima ou abaixo do dobro dessa porcentagem(Farmacopeia Brasileira, 2019).
Friabilidade
Foram empregados 20 comprimidos, os quais foram submetidos a uma pesagem precisa e, em seguida, colocados no friabilômetro. A velocidade de rotação foi ajustada para 25 rotações por minuto, durante um período total de 4 minutos. Após a remoção de qualquer resíduo em pó dos comprimidos, eles foram pesados novamente. Os comprimidos são considerados aceitáveis se apresentassem uma perda de peso igual ou inferior a 1,5% (Farmacopeia Brasileira, 2019).
Dureza
Com o auxílio de um durômetro, aparelho que mede a força aplicada necessária para esmagar o comprimido, no qual o resultado é expresso em newtons (N). Foram submetidos 10 comprimidos separadamente ao teste e a avaliação foi feita com os valores registrados e submetidos a média (Farmacopeia Brasileira, 2019).
Desintegração
Foram empregados seis comprimidos, sendo cada um deles inserido em um dos seis tubos da cesta do aparelho de desintegração. Em cada tubo, foi adicionado um disco, e utilizou-se água destilada, mantida a uma temperatura de 37 ± 1°C, como líquido de imersão. Para ser aprovado, o tempo de desintegração não pode exceder o limite máximo de 5 minutos (Farmacopeia Brasileira, 2019).
Doseamento
Foram pesados e pulverizados 20 comprimidos de AAS, transferindo uma quantidade correspondente a 0,5 g de ácido acetilsalicílico para um frasco Erlenmeyer de 250 mL, juntamente com 20 mL de solução de hidróxido de sódio 0,5 M padronizada. Após cuidadosa fervura por 10 minutos, o excesso de álcali foi titulado com solução de ácido clorídrico 0,5 M padronizada, utilizando vermelho de fenol como indicador. Um ensaio em branco foi realizado para realizar as correções necessárias. Para o cálculo da quantidade de ácido acetilsalicílico presente, foi utilizada a seguinte equivalência: cada mL de solução de hidróxido de sódio 0,5 M padronizada corresponde a 45,040 mg de ácido acetilsalicílico. A titulação foi feita em triplicata para cada laboratório e o limite deve ser no mínimo 95,0% e no máximo 105,0% de AAS (Farmacopeia Brasileira, 2019).
Dissolução
O teste de dissolução foi realizado utilizando como meio de dissolução um tampão acetato 0,05M (pH 4,5), em uma quantidade de 500 mL em cada cuba. A aparelhagem utilizada consistia em pás girando a uma velocidade de 50 rpm. Cada um dos 6 comprimidos permaneceu no dissolutor por um período de 30 minutos. Após a conclusão do teste, uma alíquota do meio de dissolução foi retirada, filtrada e diluída em tampão acetato 0,05M até atingir a concentração adequada. As absorbâncias das soluções foram imediatamente medidas em 265 nm, utilizando a solução tampão para ajustar o zero. Para calcular a quantidade de C9H8O4 dissolvido no meio, as leituras obtidas foram comparadas com as da solução padrão de ácido acetilsalicílico, preparada com uma concentração de 0,2% (p/V) no momento do uso. O padrão foi dissolvido em etanol antes da diluição em tampão acetato 0,05M. O limite de tolerância é que pelo menos 80% da quantidade declarada de C9H8O4 seja dissolvida em 30 minutos (Farmacopeia Brasileira, 2019).
RESULTADOS E DISCUSSÃO
Peso Médio
A determinação de peso é aplicada para formas farmacêuticas sólidas em dose unitárias, tal como, para comprimidos não revestidos de acordo com a Farmacopeia Brasileira 6ª e.d. (2019). Este teste é possível avaliar a uniformidade se as unidades de um mesmo lote possuem uniformidade de peso, onde será avaliado pelas variações de peso, desvio-padrão (DP) e Desvio-padrão relativo (DPR), onde é possível verificar na tabela 1.
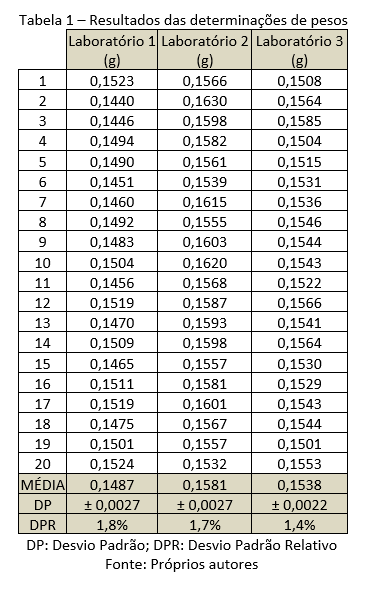
Como é descrito nesta, é aceitável tolerar não mais que duas unidades dos limites, em relação ao peso médio, e nenhuma poderá estar abaixo ou acima do dobro das porcentagens indicadas. Para os comprimidos não revestidos de ácido acetilsalicílico de 100 mg, é preconizado pelo compêndio oficial uma taxa de variação de ± 7,5%. Então as amostras dos 3 laboratórios, apresentou desvio relativo menor que 2%, sendo consequentemente aprovados.
Friabilidade
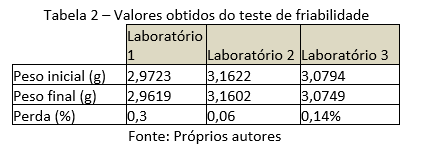
Para a análise de friabilidade segundo a farmacopeia brasileira (2019), nenhum dos comprimidos que foram submetidos devem ao final quebrar, rachar ou lascar e o limite de variação deve ser de até 1,5%. O teste se aplica unicamente a comprimidos não revestidos.
O teste traduz a resistência do comprimido ao desgaste, portanto é um parâmetro de grande importância para a verificação da perda de peso, quando submetidos a choques mecânicos decorrentes de processos industriais e de ações do cotidiano, como produção, embalagem, armazenamento, transporte e distribuição. (PEIXOTO et al.; GIL, 2007).
O laboratório 1 foi o que obteve o resultado maior de 0,3%, já o 2 e 3 obtiveram 0,06% e 0,14% respectivamente e também nenhum dos comprimidos analisados quebraram ou lascaram, portanto, ambos estão aprovados como se observa na tabela 2.
Dureza
O teste de dureza assim como a friabilidade são teste de resistência mecânica dos comprimidos, porém a dureza permite com que seja avaliada a força necessária para ruptura do mesmo. Embora o teste seja informativo ele é recomendado pela Farmacopéia Brasileira (2019).
A determinação de dureza está associada à resistência do comprimido ao esmagamento e está resistência é por sua vez, relacionada com a estabilidade física de formas sólidas obtidas por compressão. Adicionalmente, a dureza é um parâmetro essencial e imprescindível no caso de comprimidos que serão submetidos a processos de revestimento (Gil 2007)
Os resultados da dureza estão descritos na tabela 3 e expressos com a média feita em dez comprimidos de cada laboratório submetidos a análise em unidade de Newtons (N).

Desintegração
O processo de desintegração de comprimidos afeta diretamente a absorção, biodisponibilidade e ação terapêutica do fármaco. Para a ocorrência do efeito terapêutico desejado é essencial que o princípio ativo fique disponível para ser absorvido e exerça a sua ação farmacológica. Para tanto, é imprescindível ocorrer a desintegração do comprimido em pequenas partículas, aumentando assim a superfície de contato com o meio de dissolução e favorecendo absorção e a biodisponibilidade do fármaco no organismo (Peixoto et al. 2005). A velocidade de desintegração é condicionada por fatores associados ao seu processo de fabricação: compressão submetidas quantidade de desagregante utilizada (SILVA, 2019).
Resultado das análises vistas no tempo de desintegração de um fármaco reflete importantes aspectos para entender-se a ação do mesmo em organismo humano. Portanto, o limite especificado pela farmacopeia brasileira (2019) é de tempo aceitável até 5 minutos, sendo assim, os três laboratórios estão aprovados e dentro do estabelecido, apenas o laboratório 2 obteve maior tempo, tabela 4.
Observou-se que os resultados obtidos são equivalentes à literatura com tempo de desintegração 20 a 28 segundos (SILVA, 2019). A velocidade de desintegração é condicionada por fatores associados ao seu processo de fabricação: compressão submetidas quantidade de desagregante utilizada (SILVA, 2019).

Doseamento
A respeito do teor do princípio ativo presente na formulação, um dos testes que o determina é o doseamento. Este tem como importância a identificação de medicamentos com concentrações abaixo ou acima dos valores estabelecidos de qualidade, que podem acarretar intoxicação ou falta de eficácia terapêutica, prejudicando o estado clínico do indivíduo (SANTOS, 2020). Esta análise baseia-se na reação de neutralização do ácido acetilsalicílico, através da titulação e pela quantidade consumida de titulante, com isso, foi possível dosar a quantidade de AAS presente na formulação, conforme visto nos resultados apresentados na tabela 5.
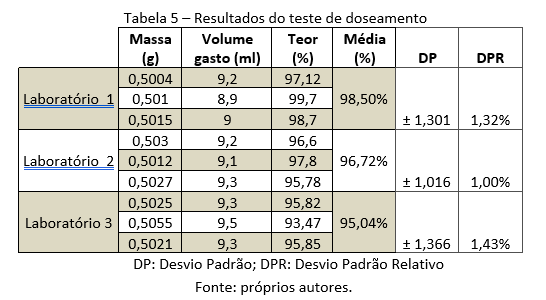
O limite preconizado pela Farmacopeia Brasileira 6 e.d. (2019) para o teste de doseamento é no mínimo 95,0% e no máximo 105,0%. Assim, podemos observar que todos os medicamentos testados obtiveram resultados dentro do esperado.
Podendo corroborar um resultado discrepante identificado por (Xavier, 2013) onde revelou que a análise de concentração do AAS em medicamentos adquiridos em farmácias e drogarias de um município no estado do Tocantins, em que todos os medicamentos estão abaixo dos padrões de qualidade, com um teor médio mínimo de 40,92% e máximo de 58,12%. Portanto, é possível que essas amostras estejam adulteradas falsificadas ou apresentem graves desvios de qualidade.
Dissolução
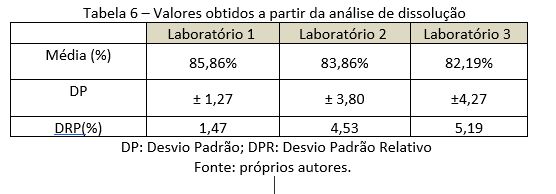
A análise de dissolução foi feita segundo as recomendações da Farmacopeia Brasileira 6ª edição (2019), em que essa medição é realizada em um único instante. Para os comprimidos de ácido acetilsalicílico, este compêndio recomenda a realização do teste de solubilidade e o limite aceitável para este medicamento é de pelo menos 80% da quantidade declarada de AAS dissolvida em 30 minutos. Isso reforça que os 2 medicamentos genéricos analisados neste artigo, possuem garantias para serem considerados intercambiáveis pelo medicamento referência, isso é regido na Lei nº 9.787, de 10 de fevereiro de 1999, que o medicamento genérico, é intercambiável pela referência, se ele apresentar um perfil de dissolução semelhante, demonstrando assim sua equivalência farmacêutica (BRASIL,1999).
Neste estudo foi conduzido, com uma única análise aos 30 minutos, o que não é suficiente para comprovar a equivalência farmacêutica. Para tal comprovação, é necessário realizar o ensaio completo do perfil de dissolução, com análises em diversos intervalos de tempo. No entanto, uma vez que o objetivo deste estudo era avaliar a qualidade dos medicamentos contendo AAS 100 mg, disponíveis para a população, e não a equivalência farmacêutica desses medicamentos, consideramos os resultados obtidos adequados, com todos os medicamentos sendo aprovados nesse teste com valores superiores a 80%, como pode ser visto na tabela 6.
CONCLUSÃO
Os testes de peso médio, friabilidade, desintegração e doseamento demonstraram que os medicamentos de referências e genéricos avaliados nesse estudo encontram-se dentro dos padrões de qualidade estabelecidos pela Farmacopeia Brasileira. Observou-se, apesar disso, que o medicamento referência apresentou maior variabilidade de massa, possuiu maior friabilidade, com perda de 0,3% de massa, porém com menor tempo de desintegração (16”), ainda deste medicamento, apresenta maior teor de AAS (98.50%) segundo o teste de doseamento. Além do mais se mostrou mais representativo em comparação aos genéricos, na sua taxa de liberação no teste de dissolução com 85.86%. Ressaltamos a importância desses testes para assegurar a qualidade e efetividade terapêutica de medicamentos. Destacamos a relevância desses testes para garantir a qualidade e eficácia terapêutica de medicamentos. Os resultados adquiridos nos permitem corroborar que a inclusão e dispersão das amostras examinadas são adequadas para o consumo.
Com base nas condições experimentais utilizadas neste estudo, conclui-se que os resultados alcançados para os comprimidos dos laboratórios testados, nos testes de doseamento, determinação de peso, dureza, friabilidade, desintegração e dissolução. Não foram observadas variações de qualidade nas várias categorias de amostras obtidas em todos os ensaios realizados, mostrando assim que os produtos disponíveis no mercado, independentemente da categoria farmacêutica, tanto os genéricos como os de referência apresentaram a qualidade desejada e recomendada pelos compêndios oficiais.
REFERÊNCIAS
CHEN, X. et al. Quality control of pharmaceutical products in China: A systematic review. PLOS ONE, v. 15, n. 11, p. e0240982, 2020.
MAIA, M. B. et al. A importância do controle de qualidade na indústria farmacêutica. Revista da SBPH, v. 19, n. 3, p. 91-97, 2016.
SILVA, A. P. et al. Farmacopeia Brasileira: uma revisão crítica sobre as tendências atuais. Revista Brasileira de Farmácia, v. 102, n. 1, p. 7-17, 2021.
NACIONAL, I. RESOLUÇÃO RDC No 658, DE 30 DE Março DE 2022 – DOU – Imprensa Nacional. Disponível em: <https://www.in.gov.br/web/dou>. Acesso em: 21 maio. 2023.
Rodrigues, T., et al. (2020). Development and validation of a dissolution test for enteric-coated aspirin tablets: an in vitro–in silico approach. Journal of Drug Delivery Science and Technology, 55, 101410.
Farmacopeia Brasileira. Disponível em: <https://www.gov.br/anvisa/pt-br/assuntos/farmacopeia/farmacopeia-brasileira/brasileira>. Acesso em: 14 jun. 2023.
GIL, E.S. Controle físico-químico de qualidade de medicamentos. 2. ed. São Paulo: Pharmabooks, 2007. 485 p.
PEIXOTO, M. M.; SANTOS JÚNIOR, A. F.; SANTOS, C. A. A.; CAETITÉ JÚNIOR, E. Avaliação da qualidade de comprimidos de captopril dispensados em Feira de Santana – BA. Infarma, v. 16, n. 13-14, 2005.
SILVA, R. P. Estudo da qualidade físico-química de comprimidos de ácido acetilsalicílico 500mg dispensados em farmácia. 36f. 2019. Monografia (graduação em Farmácia), Universidade Federal do Maranhão, São Luís, Maranhão. 2019.
XAVIER, M. P.; DE SOUSA, S. F. Análise de teor de ácido acetilsalicílico 100mg em comprimidos comercializados em farmácias e drogarias do município de Gurupi/Tocantins/Brasil. Amazônia: Science & Health, v. 1, n. 3, p. 35-42, 2013.
SANTOS, T. S., DE SOUZA, O. G. B., NETO, B. M., & DE SOUSA, P. V. A. (2020). Avaliação da qualidade de medicamentos similar, genérico e referência vendidos no Brasil: Uma revisão de literatura. Research, Society and Development, 9(7), e534974355.
BRASIL. Lei no 9.787. Dispõe sobre a vigilância sanitária, estabelece o medicamento genérico, dispõe sobre a utilização de nomes genéricos em produtos farmacêuticos e dá outras providências. Diário Oficial da União- D.O.U, 10 de fevereiro de 1999.
1Graduanda em Farmácia pelo Centro Universitário Augusto Motta (UNISUAM), Rio de Janeiro, RJ, Brasil.anapatriciasantos1415@gmail.com
2Graduando em Farmácia pelo Centro Universitário Augusto Motta (UNISUAM), Rio de Janeiro, RJ, Brasil.matheusgomescontatos@gmail.com
3Mestre em Ciências pela Universidade Federal Fluminense, UFF. Farmacêutico da Atenção Básica no Município do Rio de Janeiro e Docente do curso de Farmácia do Centro Universitário Augusto Motta, UNISUAM, Rio de Janeiro, RJ. E-mail: darlanferreira@souunisuam.com.br Autor para correspondência