REGISTRO DOI: 10.5281/zenodo.7603107
Alexandre Dal’Maso1
Gustavo Henrique Dalposso1
Priscila Debastiani Barros2
Resumo
A estratégia adotada neste trabalho foi avaliar o impacto da interrupção de uma produção em campanha por tempo determinado, com a retomada de produção até a conclusão do número total de lotes previstos, e com isso, evidenciar que o período de interrupção não promove possíveis contaminantes para os lotes envolvidos no processo. O estudo foi realizado em uma linha de embalagem de comprimidos em uma empresa farmacêutica da região oeste paranaense. Foi identificado o produto mais crítico para a execução da validação de limpeza e foi definido que seriam analisadas três campanhas produtivas, sendo cada uma delas composta por vinte lotes, com tempo de parada dentro da campanha produtiva de no mínimo quarenta e oito horas. Foram realizadas coletas nos tempos e nos pontos pré-determinados e as mesmas foram analisadas quanto à detecção de resíduos da substância ativa, presença de microrganismos e a presença de produtos de degradação. Após a produção dos lotes foi possível identificar o tempo de interrupção padrão entre as campanhas e o tempo total (em dia) de duração de uma campanha. Os resíduos dos possíveis contaminantes no equipamento após a limpeza geral foram inferiores aos limites dos critérios de aceitação admitidos para o estudo. Mesmo com a interrupção do processo, o emprego da produção em campanha e o tempo total de processo, o procedimento de limpeza estabelecido foi eficiente quanto a remoção dos resíduos microbiológicos, resíduos do medicamento, e possível geração de substâncias de degradação durante o processo.
Palavras-chave: validação de limpeza; produção em campanha; boas práticas de fabricação; indústria farmacêutica; contaminação cruzada.
Abstract
The strategy adopted in this work was to evaluate the impact of the interruption of a production campaign for a determined period of time, with the resumption of production until the completion of the total number of batches planned, and thereby demonstrate that the interruption period does not promote possible contaminants for the batches involved in the process. The study was conducted in a tablet packaging line in a pharmaceutical company in the western region of Paraná. The most critical product was identified for the execution of the cleaning validation and it was defined that three production campaigns would be analyzed, each one of them composed of twenty lots, with stoppage time within the production campaign of at least forty-eight hours. Samples were collected at the predetermined times and points and were analyzed for residue detection of the active substance, presence of microorganisms and the presence of degradation products. After the production of the batches it was possible to identify the standard interruption time between the campaigns and the total time (in days) of duration of a campaign. The residues of possible contaminants in the equipment after the general cleaning were below the limits of the acceptance criteria allowed for the study. Even with the process interruption, the campaign production and the total process time, the established cleaning procedure was efficient in removing microbiological residues, drug residues, and possible generation of degradation substances during the process.
Keywords: cleaning validation; campaign manufacturing; Good Manufacturing Practices; pharmaceutical industry; cross contamination.
Introdução
A validação de limpeza é a evidência documentada que um procedimento de limpeza utilizado em um determinado equipamento é eficaz e reprodutível quanto a remoção dos resíduos resultantes da produção de lotes de medicamentos e resíduos de agentes de limpeza. Ainda, promove a redução da carga microbiana à níveis cientificamente aceitáveis, avalia a ocorrência de formação de substâncias tóxicas durante o processo produtivo (1), protege a integridade do produto, possibilita a reutilização dos equipamentos nos processos, bem como a necessidade em se cumprir com as exigências das autoridades reguladoras (2).
A validação de limpeza garante a implementação de um procedimento eficiente de limpeza que previne a “contaminação cruzada” entre produtos diferentes ou lotes diferentes do mesmo produto (3). A preocupação com possibilidade das contaminações cruzadas foi descrita em 1963, quando o órgão americano Food and Drug Administration (FDA) determinou que os equipamentos devem ser mantidos de forma limpa e ordenada (4). Como forma de evitar a contaminação por resíduos de produtos diferentes em um mesmo equipamento, em 1978 o FDA publicou que os equipamentos e utensílios devem ser mantidos limpos e higienizados em intervalos apropriados para evitar contaminação e procedimentos escritos devem ser estabelecidos e seguidos para a limpeza e manutenção do equipamento (5).
A validação de limpeza garante que os resíduos remanescentes do lote produtivo tenham sido retirados, a níveis aceitáveis, durante o processo de limpeza. Observa-se, através de monitoramento analítico, a remoção dos produtos residuais, produtos de degradação, conservantes, excipientes e/ou agentes de limpeza de modo a obedecer às especificações e limites de cada um desses (6).
Uma das características principais da validação de limpeza é que ela envolve tanto o produto finalizado quanto o próximo produto a ser fabricado no equipamento já limpo (7). Logo, a sequência com que se fabricam os produtos nestas unidades, influencia a validação de limpeza da unidade multipropósito. Dessa forma, o estudo de determinação do pior caso deve ser baseado em parâmetros como solubilidade e toxidade do produto ativo, menor tamanho de lote que pode ser fabricado com o equipamento em questão, a dose máxima diária deste produto e o número de dosagens que podem ser feitas do próximo lote. A avaliação destes fatores proporciona o índice WCI (Worst Case Index), que determina qual o medicamento, dentro de um grupo de produtos fabricados em unidade fabril multipropósito, será utilizado como o pior caso.
A produção em campanha é uma estratégia adotada nas indústrias farmacêuticas com o objetivo de obter eficiência e otimização nas produções dos medicamentos. Uma estratégia de resultados satisfatórios, porém já reforçada pelo FDA, é que, o número de lotes e tempo máximo da campanha são determinados pela empresa durante o estudo de validação de limpeza (8).
LeBlanc (9) publicou em seus memorandos, a existência de pelo menos três tipos diferentes de campanhas, sendo:
Caso I: uma campanha na qual existe uma limpeza totalmente validada entre cada lote na campanha;
Caso II: uma campanha na qual existe algum tipo de “limpeza menor” entre cada lote, e somente uma limpeza geral, totalmente validada, no final de um determinado número de lotes ou após um certo tempo decorrido ou no final de uma campanha produtiva.
Caso III: uma campanha na qual não há limpeza de qualquer tipo entre cada lote, mas apenas uma limpeza totalmente validada no final de um certo número de lotes ou após um certo tempo decorrido (ou no final da campanha produtiva).
Na Instrução Normativa (IN) Nº 47 de 2019 a ANVISA (Agência Nacional de Vigilância Sanitária) é previsto que quando a fabricação da campanha é realizada, o impacto na facilidade da limpeza ao final da campanha deve ser considerado e a duração máxima da campanha (em tempo e número de lotes) deve ser a base para os exercícios de validação de limpeza (10).
Neste trabalho, propôs-se a avaliação da eficiência do procedimento de limpeza de uma emblistadeira de comprimidos na condição de produção de três campanhas produtivas, cada uma constituída de 20 lotes do produto crítico com o tempo de parada de no mínimo 48 horas após o 15º lote de cada campanha, sem limpeza ao retornar a produção. O processo foi avaliado quanto a remoção dos resíduos promissores de contaminação cruzada, como as substâncias ativas de lotes anteriores, contaminantes microbiológicos, presença de substâncias de degradação durante o processo, além de determinar o tempo de campanha e parada padrão para implementação na rotina da empresa.
Material e métodos
Delineamento do estudo
A execução da proposta ocorreu na área de produção de medicamentos da forma farmacêutica sólidos orais. O equipamento determinado para avaliação foi a emblistadeira de comprimidos, (marca ACG-pampac®, modelo Cartoblis Compact Drive Machine) onde ocorre a etapa do emblistamento, ou seja, o acondicionamento do produto na sua embalagem primária (Blíster). Foram produzidos três campanhas produtivas de vinte lotes do produto de escolha (produto crítico). A interrupção proposta para o estudo foi realizada após o 15º lote, com o tempo mínimo de 48 horas. A produção do 16º lote foi retomada sem a execução de qualquer tipo de limpeza, tanto no equipamento como na área física. A produção de cada campanha foi conduzida até o término da produção do 20º lote. Como critério para padronização do tempo de produção, foram utilizados os tempos que se repetiram dentro de cada campanha.
Foram coletadas as amostras para análises de contaminantes microbiológicos, no tempo inicial da parada, representado por T0, e após a espera total da parada pré-estabelecida antes de iniciar a retomada da produção da campanha, indicado por T1 (Figura 1).
Após o último lote da campanha, a partir da finalização da limpeza (T2), fez-se o processo da avaliação visual no equipamento e as coletas conforme os pontos de amostragens para quantificar a presença de contaminantes microbiológicos (P01, P02 e P03 – RODACTM JProlab®, P04, P05 e P06 – swab estéril Neolab®), presença de resíduos de ativo (swab por esfregaço Texwip® umedecidos em solução de ácido acético Synth®) e detecção de produtos de degradação (Coleta de comprimidos) (Figura 2).
Para avaliação da eficiência da limpeza geral, foi utilizado o procedimento operacional padrão (POP) que é aplicado após a produção de medicamentos nos equipamentos de emblistamento de comprimidos. O POP avaliado tem como principais etapas de execução de limpeza: desmontagem do equipamento, lavagem das partes e peças com detergente neutro (Diversey®), enxague utilizando água potável e purificada, e por fim a sanitização com álcool 70% (Emfal®).
Determinação do produto alvo do estudo
Para a definição do produto a fazer parte do estudo, calculou-se o WCI (Worst Case Index) de cada um dos 147 produtos da empresa que utilizam a emblistadeira, e o produto de escolha (pior caso) foi aquele que apresentou o maior índice WCI. A Equação (1) representa o cálculo do WCI, que será expresso em valor numérico, quanto maior o valor, será indicado o produto considerado o pior caso, dada pela equação:
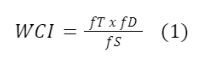
Onde: fT: Fator de Toxidade; fD: Fator de Dificuldade de Limpeza e fS: Fator de solubilidade.
O Fator de Toxidade (fT) foi obtido pela comparação da toxicidade do medicamento com o valor da DL50 (Dose Letal – o grau de toxicidade aguda de um produto). Os critérios para determinação do fT foram determinados conforme Guia de Validação de limpeza para farmoquímicas da ANVISA e no estudo de Klaassen (11, 12). O fator de dificuldade de limpeza (fD) é fornecido pelos operadores envolvidos nos processos de limpeza dos equipamentos, expondo a experiência na execução das atividades de limpeza. O fator de Solubilidade (fS), é a capacidade de uma determinada substância ser dissolvida em um solvente conhecido ou que será utilizado no processo de limpeza, conforme literatura disponível (11, 13).
Determinação microbiológica
Para determinação de bactérias, aplicou-se uma alíquota das amostras coletadas com swab estéril em placa de Petri, adicionou-se 20 mL de Ágar Caso (Merck®) e incubou-se em estufa (marca Panasonic®, modelo MIR-154-PA) na temperatura de 30 a 35 °C por 48 horas, juntamente com as placas de Rodac contendo Ágar Caso (JProlab®). Para determinação de bolores e leveduras, aplicou-se uma alíquota das amostras coletadas com swab estéril em placa de Petri, adicionou-se 20 mL de Ágar Caso, e incubou-se em estufa na temperatura de 20 a 25 °C por 72 horas, juntamente com as placas de Rodac contendo Sabouraud (JProlab ®). Para a identificação de Escherichia coli, utilizou-se a técnica de coloração de gram para bacilo gram negativo, coletou-se uma das colônias do Ágar Caso, estriou-se na superfície de Ágar MacConkey (Merck®) e a mesma foi incubada em estufa na temperatura de 30 °C a 35 °C por 18 a 72 horas. Conforme metodologia desenvolvida pela empresa, os limites de aceitação para bactérias é de no máximo 50 UFC/25cm², para fungos e leveduras de no máximo 6 UFC/25cm² e para patógenos deverá ser ausente (14).
Determinação de Resíduo de substância ativa
Foi amostrado ao término da produção do 20º lote de cada campanha produtiva, após a realização da limpeza geral no equipamento emblistadeira de comprimidos, no tempo T2. Para a quantificação do resíduo de substância ativa do medicamento utilizou-se a técnica de doseamento por Cromatografia Líquida de alta Eficiência (HPLC – Equipamento marca Shimadzu®, modelo LC-2030C 3D), na configuração e utilização da solução Fosfórico 0,01M (Scharlau®), em gradiente de 50:50 com Acetronitrila (Biograde®) grau HPLC, em leituras em UV 220 nm, corridas com 18 minutos e tempo de retenção em 9,5 minutos, conforme metodologia desenvolvida pela empresa.
Para determinar o limite de aceitação para quantificação dos resíduos de substância ativa, a empresa realizou os cálculos do limite de resíduos de ativo utilizando a metodologia de cálculo de limite de aceitação conforme indicado por Leblanc (9) e metodologia estabelecida no Guia de Validação de Limpeza para Farmoquímicas (11).
Diante dos cálculos obtidos, utilizando do Limite do pior caso no Produto Subsequente (LPS), o Limite por Área Superficial (LAS) e Limite na Amostra Analisada (LAA), resultou no limite de aceitação de 0,855 ppm, desta forma, resultados encontrados acima deste valor serão insatisfatórios.
Determinação de produtos de degradação
Foi realizada a técnica de quantificação do composto 4-isobutilacetofenona por HPLC (Equipamento marca Shimadzu®, modelo LC-2030C 3D). Para cada lote produzido, triturou-se a fino pó 10 comprimidos do medicamento, preparou-se em solução de fase móvel de ácido tricloacético (Sigma Aldrich®) e Acetronitrila 40:60 (Biograde®), para aplicar no sistema cromatográfico, em leitura de comprimento de onda UV 254 nm, em corrida de 10 minutos com tempo de retenção próximo a 6,8 minutos. A empresa estabeleceu o limite de aceitação para o produto de degradação a partir de desenvolvimento local seguindo as diretrizes da Resolução RDC Nº53/2015 ANVISA (15) determinando que os resultados encontrados não podem ser mais de 10% da substância.
Análise de Inspeção Visual
Ao concluir o processo de limpeza geral do equipamento emblistadeira de comprimidos, após a produção de cada campanha produtiva, composta de 20 lotes do medicamento, foi executada a inspeção visual.
A avaliação ocorreu observando de forma direta os pontos de amostragem em cada parte e peças que estiveram em contato com produto ao final de cada campanha produtiva, verificando a presença de partículas, ou seja, resíduos possíveis de serem observados ao nível de detecção aos olhos.
Resultados
Delineamento do estudo
Após a produção das três campanhas do medicamento, cada uma constituída por 20 lotes, foi possível obter o tempo de parada entre o 15º e 16º lote de cada campanha, e também o tempo total da campanha em dias, conforme descrito na Tabela 1. Os resultados da eficácia do procedimento de limpeza e das amostragens estarão descritos nos itens subsequentes.
Determinação do produto alvo do estudo
Com os critérios de fT, fD e fS, foi construída a matriz contendo todos os medicamentos que são produzidos na linha produtiva que utiliza o equipamento emblistadeira de comprimidos. Ao empregar a o cálculo conforme a Equação (1), o maior resultado obtido do índice WCI foi o Medicamento 3, com o valor de 9,00, sendo este o produto escolhido para o estudo.
Determinação microbiológica
Na avaliação de atividades dos microrganismos nos pontos amostrados, todos os resultados (Tabela 2) apresentaram dentro dos critérios de aceitação, assim demonstrando que os resultados são satisfatórios no cumprimento desta etapa de avaliação para a Validação de Limpeza.
Determinação de Resíduo de substância ativa
Os resultados da verificação da presença de resíduos da substância ativa são apresentados na Tabela 3. Verifica-se que em todos os pontos amostrados, os critérios de aceitação com valores abaixo de 0,85 ppm foram atendidos.
Os resultados analíticos de detecção da substância ativa em cada campanha podem ser evidenciados nos cromatogramas, apresentando a relação entre os resultados analíticos dos padrões e amostras analisadas. A Figura 3 demonstra os resultados encontrados ao final da 1ª campanha produtiva. Na Figura 3(A) observa-se o pico da substância ativa do medicamento de concentração conhecida, valor de 2,410 ppm, valor determinado por metodologia analítica empregada pela empresa. Na Figura 3(B) estão representadas as corridas analíticas para todos os pontos de coleta, do ponto P1 ao P6. Os pontos P02, P03 e P04, apresentam picos nos cromatogramas, indicando a presença da substância ativa nas amostras, valores de 0,451, 0,269 e 0,104 ppm, respectivamente.
A Figura 4 apresenta os resultados encontrados ao final da 2ª campanha produtiva. Na Figura 4(B), observa-se que os pontos P02 e P03 apresentam picos nos cromatogramas, demonstrando a presença da substância ativa nas amostras, de 0,310 e 0,105 ppm respectivamente.
Nos resultados encontrados na 3ª campanha produtiva, observa-se que foi encontrado 0,590 ppm da substância ativa na amostra do ponto P02 (Figura 5(B)).
Determinação de produtos de degradação
Estão reportados na Tabela 4 os resultados de detecção da substância 4-isobutilacetofenona em cada lote das três campanhas produtivas empregadas no presente estudo. As análises foram satisfatórias pois atenderam a especificação de não mais que 0,10% da substância presente na amostra coletada.
Análise de Inspeção Visual
A Tabela 5 apresenta a avalição qualitativa realizado no estudo. Verifica-se que todos os pontos analisados atenderam os critérios de aceitação para a inspeção visual, ou seja, sem presença de sujidade visível aos olhos.
Discussão
Delineamento do estudo
A abordagem deste trabalho baseou-se no estudo de Caso II apresentado por LeBlanc (9) onde o mesmo define que, mesmo que exista uma “limpeza menor” (limpeza parcial) entre os lotes, a validação de limpeza é concluída somente ao final de um determinado número de lote (produção em campanha) ou após um certo tempo decorrido da produção de todos os lotes constituintes da campanha. Dessa forma, a escolha da interrupção do processo no 15º lote justifica-se pelo fato de que a produção deste lote coincide exatamente com o final de semana, onde ocorrem as paradas produtivas na indústria, proporcionando o melhor aproveitamento do tempo produtivo durante o período de trabalho semanal, sem interrupções no mesmo.
Diante dos resultados da Tabela 1, observou-se que mesmo que as campanhas tenham sido conduzidas na mesma configuração de produção, é esperado que os tempos de execução de cada campanha sejam influenciados diretamente pela programação de produção de cada lote, e também, a condição e disponibilidade das equipes em executar os processos de produção.
Além de padronizar a quantidade de lotes de cada campanha, com os resultados dos tempos, poderemos indicar o tempo padrão da produção em campanha produtiva na rotina. Ao comparar os resultados do tempo de interrupção em uma campanha, observou-se que o tempo de 50 horas foi comum entre as os estudos. No cumprimento das legislações vigentes, conforme a IN Nº47, deve ser definido o número de lotes e o tempo máximo para produção de lotes em campanha (10). Podemos observar que o tempo total da 2ª campanha foi o menor tempo encontrado, no qual está contemplado desde o início da produção do 1º lote, com o tempo de interrupção e retorno da produção da campanha, até o término da produção do 20º lote, com o resultado equivalente a 9,08 dias de produção. Com maior tempo total, observa-se a 1ª campanha, apresentando resultados de 12,57 dias de produção. Conforme o critério adotado, iremos padronizar como tempo total o tempo que pôde ser observado em todas as campanhas, desta forma, para o tempo total padrão foi estabelecido o período de 9,08 dias de produção em campanha.
Para realizar a avaliação da eficiência do procedimento de limpeza, foram realizadas amostragens na superfície do equipamento, nas partes e peças que entraram em contato direto como o produto alvo do estudo. O processo de amostragem ocorreu em dois momentos do estudo, primeiramente durante a parada da campanha, o segundo momento ocorreu após a conclusão da limpeza geral do equipamento.
O princípio da escolha dos pontos de amostragem foi baseado na avaliação do contato entre partes do equipamento com o medicamento alvo do estudo, ainda, que o processo de amostragem fosse capaz de reter os possíveis contaminantes e os resíduos dos componentes, caso estivessem presentes nas superfícies do equipamento (3). A relevância dos pontos de coleta foi demonstrada pelos estudos pioneiros de Leblanc (2) que citaram a inclusão da amostragem em locais onde a limpeza seja feita com maior dificuldade ou onde se tenha o maior acúmulo de resíduo (válvulas de entrada e saída, conexões), pontos que representem a função dos equipamentos (paredes, agitadores), pontos que possam produzir contaminações nos próximos produtos (agulhas, bicos dosadores).
Determinação do produto alvo do estudo
A escolha do produto alvo do estudo é determinada pela avaliação de fatores que mensuram atributos dos medicamentos que são produzidos na linha produtiva e a condição de limpeza no equipamento. Neste estudo, o maior valor obtido neste índice, foi do Medicamento 3, dentro de um grupo de produtos fabricados em unidade fabril multipropósito, e o mesmo utilizado como o pior caso para o estudo de Validação de Limpeza. A determinação do pior caso, conforme explica Nassani (16), é uma etapa crucial na definição dos limites de contaminação e eficácia do procedimento de limpeza.
Determinação microbiológica
Para a avaliação dos resultados microbiológicos, buscou-se identificar se durante os tempos de exposição de vários lotes utilizando o mesmo equipamento, ocorreria a proliferação microbiana, para bactérias, bolores e leveduras. Todos os valores apresentarem-se inferiores a 50 UFC/cm², porém, como as leituras das análises não tiveram a presença de microrganismos viáveis para formação de colônias microbianas, não foi realizado a contagem UFC (Unidade Formadora de Colônia). Desta forma, os resultados foram expressos como menores que 6 UFC/25cm².
Na avaliação de patógenos foi analisado a presença de Escherichia coli (Tabela 2). De acordo com os resultados, observou-se que todos as amostras ficaram dentro do especificado, pois foi constatado a ausência deste microrganismo em todos os pontos executados nas três campanhas.
Para análise microbiológica, nos pontos de P01, P02 e P03, foi realizada a coleta das amostras utilizando as placas de RODACTM. Essa metodologia é indicada em virtude das superfícies dos pontos serem planas. Nos pontos P04, P05 e P06, foram utilizados amostradores tipo swab.
A Farmacopeia Brasileira 6ª edição (17) descreve a necessidade de executar o controle biológico, identificando a flora microbiana e as bactérias patogênicas, estabelece ainda que, em resultados acimas dos níveis aceitáveis os microrganismos podem ser prejudiciais aos processos e produtos. Esta ocorrência pode acarretar a altos custos de descartes de produtos, grandes esforços para eliminação dos contaminantes e promover risco sanitário aos consumidores dos medicamentos contaminados.
É importante salientar que na condução do desenho do estudo, o T0 apresentou resultados satisfatórios para as análises microbiológicas, sendo que a coleta foi realizada após a conclusão do 15º lote. Antes da realização da coleta, foi realizada somente a retirada dos materiais do lote da sala produtiva, e o equipamento não sofreu qualquer tipo de limpeza. Ainda, após a execução dos tempos de interrupção da campanha, foi realizada a coleta do tempo T1, sendo que neste momento o equipamento permaneceu conforme condições deixadas antes da coleta do tempo T0. Os experimentos demonstram que o tempo de parada de 50 horas não possibilitou a proliferação de microorganismos e /ou bolores e leveduras, preservando a qualidade do medicamento, e permitindo a flexibilização da programação de produção.
Determinação de Resíduo de substância ativa
De acordo com os resultados analíticos via metodologia por HPLC, avaliamos que os dados atendem ao critério de aceitação, sendo assim satisfatório, demonstrando que o procedimento de limpeza é eficaz na remoção da substância ativa do pior caso empregado no estudo.
No estudo de Hall (18) foram descritos os principais elementos para validar um processo de limpeza, sendo um deles o procedimento escrito que determine o nível de limpeza desejado e os métodos de ensaios analíticos com sensibilidade adequada para o teste de determinação de níveis de aceitáveis residuais.
Determinação de produtos de degradação
Observa-se que os resultados não superaram o limite da aceitação para a substância química 4-isobutilacetofenona. Desta forma, tem-se a indicação que durante a produção em campanha produtiva, mediante a condição de execução do estudo, os lotes avaliados (1º lote ao 20º lote), referentes a 1ª, 2ª e 3ª campanhas, apresentaram resíduos de produto de degradação abaixo do valor do critério de aceitação.
Moretto e Calixto (19), descrevem que é de extrema importância que a pesquisa de produtos de degradação seja realizada, pois essas substâncias podem surgir durante a limpeza e representar riscos devido a sua toxicidade. Por esse motivo, os estudos de produtos de degradação encontram-se em uma esfera de grande discussão entre os órgãos reguladores e indústrias farmacêuticas (20).
Análise de Inspeção Visual
A inspeção visual foi conduzida para investigar cada ponto de amostragem, quanto a presença de partículas visíveis, possíveis de serem visualizadas pelo responsável da avaliação. LeBlanc (2) descreve que, o ponto para a avaliação da inspeção visual, é que se uma superfície estiver visualmente suja, então o procedimento de limpeza não é aceitável ou uma vez aceitável o procedimento está fora de controle.
Mesmo sendo uma análise qualitativa e que pode ser influenciada pela da subjetividade do analista que a executa, Andrade (21) demonstrou resultados satisfatórios em um estudo de validação de limpeza para equipamentos farmacêuticos com a análise visual. Este estudo indica que a inspeção visual pode ser aplicada nas validações da indústria farmacêutica. Porém, ainda que todos os resultados da inspeção visual sejam conforme, atualmente observa-se que no aspecto de execução e manutenção de um processo de validação de limpeza na indústria atualizado e robusto, é fundamental que seja aplicado outros métodos capazes de demonstrar que o processo de limpeza seja eficaz atendendo os critérios de aceitação (2, 10).
Conclusões
Este estudo demonstrou que a produção em campanha de 20 lotes de medicamento com tempo de interrupção de 50 horas foi adequada para ser implementado no equipamento de emblistamento de comprimidos. Foi evidenciado que, mesmo com a interrupção do processo, o emprego da produção em campanha e o tempo de processo acumulado (tempo decorrido do início da produção do primeiro lote da campanha até o último) o processo de limpeza estabelecido ainda é eficiente quanto a remoção dos resíduos promissores de contaminação cruzada, como as substâncias ativas de lotes anteriores, contaminantes microbiológicos e possível geração de substâncias de degradação durante o processo. Além dos resultados que demonstram segurança e qualidade na execução da produção em campanha, é possível que com essas estratégias, possa ocorrer o ganho produtivo no processo, como a diminuição dos números de paradas para limpezas gerais e diminuição dos custos operacionais. Desta forma, este trabalho demonstra a possibilidade da realização de novos ensaios para evidenciar os possíveis ganhos produtivos.
Referências
1. BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RDC nº 301, de 21 de agosto de 2019. Dispõe sobre as Diretrizes Gerais de Boas Práticas de Fabricação de Medicamentos. Diário Oficial da União, nº 162, 22 de agosto de 2019. Seção 1, p 64.
2. Leblanc, DA. Validated cleaning technologies for pharmaceutical manufacturing. Interpharm Press. Boca Raton: CRC Press, 2000.
3. Haider, SI, Asif, SA. Cleaning Validation Manual. A Comprehensive Guide for the Pharmaceutical and Biotechnology Industries. Boca Raton: CRC Press, 2010.
4. FDA. U.S. Food & Drug Adminitration. Validation of clean Proceses (7/93) – Guide to Inspections Validation of Cleaning Processes. USA, 2014.
5. FDA. U.S. Food & Drug Adminitration. Current Good Manufacturing Practice in Manufacture, Processing, Packing, or Holding. USA, 43(190), 1978.
6. Tavares, SA. Pichatelli, FP, Prado, MCGB. A importância da validação de limpeza na indústria Farmacêutica: Revisão de Literatura. Revista Científica Multidisciplinar Núcleo do Conhecimento. 2018; 02(01): 85-100.
7. Alencar, JRB, Clementino, MRA, Neto, PJR. Validação da limpeza de equipamentos numa indústria de medicamentos: estratégia para escolha do pior caso. Revista Brasileira Farmacêutica. 2006; 87(1):13-18.
8. FDA. U.S. Food & Drug Adminitration. Guide to inspections of Validation of Cleaning Processes. Division of Investigations. Office of Regional Operations. Office of Regulatory Affairs. USA, 1993.
9. Leblanc, D.A. Public the of Memos: Issues in Campaigns. Quascenta – Cleaning Validation Technologies. June, 2007.
10. BRASIL. Agência Nacional de Vigilância Sanitária. Instrução Normativa – IN nº 47 de 21 de agosto de 2019. Dispõe sobre as Boas Práticas de Fabricação complementares às atividades de qualificação e validação. Diário Oficial da União, nº 192, 22 de agosto de 2019. Seção 1. P.96.
11. BRASIL. Agência Nacional de Vigilância Sanitária. Guia de Validação de limpeza para farmoquímicas. Brasília, 2013.
12. Klaassen, CD. Casarett & Doull’s Toxicology, the basic science of poisons. USA: The McGraw-Hill Companies; 2008.
13. United States Pharmacopeia, 41st ed. USP convention, 2021.
14. Leblanc, DA. Equipment Cleaning Validation: Microbial Control Issues. Journal of Validation Technology; 2002; 8(4):40-46.
15. BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RDC nº 53 de 4 de dezembro de 2015. Estabelece parâmetros para a notificação, identificação e qualificação de produtos de degradação em medicamentos com substâncias ativas sintéticas e semissintéticas, classificados como novos, genéricos e similares, e dá outras providências. Diária Oficial da União, nº 234, de 8 de dezembro de 2015. Seção 1, páginas 48 e 49.
16. Nassani, M. Cleaning Validation in the Pharmaceutical Industry. J Vali. 2005;38-58.
17. BRASIL. Agência Nacional de Vigilância Sanitária. Resolução. Farmacopeia Brasileira 6ª Edição. Volume 1. Resolução RDC Nº 298, 12 de agosto de 2019. Dispõe sobre a aprovação da Farmacopeia Brasileira. Diário Oficial da União, nº 156, 14 de agosto de 2019. Seção 1. p. 63.
18. Hall, WE. Pharmaceutical process validation: An international third edition, revised and expanded. Drugs and the pharmaceutical sciences. New York; 2003. 14, Validation and Verification of Cleaning Processes.
19. Moretto, LD, Calixto, J. Qualificações e Validações: Guia de Qualificação e validação. SINDUSFARMA. Manuais SINDUSFARMA, v. 17. São Paulo: Câmara Brasileira do Livro; 2013.
20. FARIAS, FF. Estudo de degradação forçada e caracterização das principais impurezas da cápsula líquida de ibuprofeno por LC-MS-QTOF. (Dissertação mestrado) São Paulo: USP – Faculdade de Ciências Farmacêuticas; 2020.
21. ANDRADE, SCI. Validação de limpeza de equipamentos farmacêuticos. (Dissertação Mestrado), Portugal: Instituto Politécnico de Leiria, Escola Superior de Turismo e Tecnologia do Mar; 2012.
Figura 1. Representação da proposta para condução da produção de lotes em campanhas com interrupção durante o processo produtivo.

Figura 2. Esquema dos pontos de amostragem para realização das coletas para as análises analíticas. P01: funil de alimentação; P02: canaleta de descida dos comprimidos na bandeja vibratória; P03: centro da bandeja vibratória; P04: direcionador de comprimidos; P05: parte orientadora dos comprimidos para a calha de alimentação; P06: calha de alimentação.

Tabela 1. Tempos encontrados durante a produção das três campanhas produtivas.
Tempos na Campanha Produtiva 1ª Campanha 2ª Campanha 3ª Campanha Tempo de Parada (Horas) 61,83 50,33 50,00 Tempo total da campanha (Dias) 12,57 9,08 10,76
Tabela 2. Resultados das análises dos contaminantes microbiológicos das campanhas produtivas.
Pontos 1ª Campanha 2ª Campanha 3ª Campanha T0 T1 T2 T0 T1 T2 T0 T1 T2 P01 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 A A A A A A A A A P02 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 A A A A A A A A A P03 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 A A A A A A A A A P04 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 A A A A A A A A A P05 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 A A A A A A A A A P06 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 ≤6 A A A A A A A A A
T0: Tempo zero; T1: Tempo 1; T2: Tempo 2; P01: funil de alimentação; P02: canaleta de descida dos comprimidos na bandeja vibratória; P03: centro da bandeja vibratória; P04: direcionador de comprimidos; P05: parte orientadora dos comprimidos para a calha de alimentação; P06: calha de alimentação; * Quando o resultado encontrado for de 0 UFC/25cm², o resultado será expresso como <6UFC/25cm². A – Ausente.
Tabela 3. Resultados analítico da detecção da concentração de resíduos da substância ativa.
Pontos 1ª Campanha 2ª Campanha 3ª Campanha Tempo 2 (T2) Tempo 2 (T2) Tempo 2 (T2) P01 0,000 0,000 0,000 P02 0,451 0,310 0,590 P03 0,269 0,105 0,000 P04 0,104 0,000 0,000 P05 0,000 0,000 0,000 P06 0,000 0,000 0,000
O Parâmetro de aceitação para a presença de resíduo da substância ativa deve ser menor que 0,85 ppm. P01: funil de alimentação; P02: canaleta de descida dos comprimidos na bandeja vibratória; P03: centro da bandeja vibratória; P04: direcionador de comprimidos; P05: parte orientadora dos comprimidos para a calha de alimentação; P06: calha de alimentação.
Figura 3. Cromatogramas das análises analíticas por HPLC das amostras referente a 1ª Campanha produtiva. (A): Resultado da amostra do padrão analítico e (B): Resultados das amostras coletadas no equipamento.

Figura 4. Cromatogramas das análises analíticas por HPLC das amostras referente a 2ª Campanha produtiva. (A): Resultado da amostra do padrão analítico e (B): Resultados das amostras coletadas no equipamento.

Figura 5. Cromatogramas das análises analíticas por HPLC das amostras referente a 3ª Campanha produtiva. (A): Resultado da amostra do padrão analítico e (B): Resultados das amostras coletadas no equipamento.

Tabela 4. Resultados analíticos da detecção dos limites 4-isobutilacetofeno dos lotes produtivos, critério de aceitação não mais que 0,10%.
Lotes Produto e Degradação não mais que 0,10% 1ª Campanha 2ª Campanha 3ª Campanha Lote1 0,00 0,00 0,00 Lote2 0,00 0,00 0,00 Lote3 0,00 0,00 0,00 Lote4 0,00 0,00 0,00 Lote5 0,00 0,00 0,00 Lote6 0,00 0,00 0,00 Lote7 0,00 0,00 0,00 Lote8 0,00 0,00 0,00 Lote9 0,00 0,00 0,00 Lote10 0,00 0,00 0,00 Lote11 0,00 0,00 0,00 Lote12 0,00 0,00 0,00 Lote13 0,00 0,00 0,00 Lote14 0,00 0,00 0,00 Lote15 0,00 0,00 0,00 Lote16 0,00 0,00 0,00 Lote17 0,00 0,00 0,00 Lote18 0,00 0,00 0,00 Lote19 0,00 0,00 0,00 Lote20 0,00 0,00 0,00
Tabela 5. Resultados da determinação da inspeção visual após a realização da limpeza geral de cada campanha produtiva.
Pontos 1ª Campanha 2ª Campanha 3ª Campanha Tempo 2 (T2) Tempo 2 (T2) Tempo 2 (T2) P01 C C C P02 C C C P03 C C C P04 C C C P05 C C C P06 C C C
C: Conforme; T2: Tempo 2; P01: funil de alimentação; P02: canaleta de descida dos comprimidos na bandeja vibratória; P03: centro da bandeja vibratória; P04: direcionador de comprimidos; P05: parte orientadora dos comprimidos para a calha de alimentação; P06: calha de alimentação.
1Universidade Tecnológica Federal do Paraná. Rua Cristo Rei, 19, CEP 85902-490. Toledo, PR, Brasil.
2Universidade Tecnológica Federal do Paraná. Rua Cristo Rei, 19, CEP 85902-490. Toledo, PR, Brasil.