BILIARY ATRESIA AS A CAUSE OF OBSTRUCTIVE JAUNDICE
REGISTRO DOI: 10.69849/revistaft/ra10202503092321
ARRUDA, Adriana Lima Resende1
ARRUDA, Ana Júlia Resende2
CHICUMBI, John Alex Pereira3
DA ROCHA, Ana Percya Sais4
DOS SANTOS, Jacqueline Ramos5
FILHO, André Bernardes Silva6
GOMES, Júlio Cesar de Oliveira7
HUL, Stefan Fausto8
PACHECO, Vilson Damião Silva9
XAVIER, Thayse Ventura10
RESUMEN
La atresia biliar (AB) es una condición grave, idiopática y progresiva, con una incidencia mundial de 1 por cada 8,000 a 15,000 nacidos vivos, siendo más común en países asiáticos. La enfermedad provoca hiperbilirrubinemia, heces acólicas, orina oscura, además de hepatomegalia y esplenomegalia, y, si no tratada, puede llevar a la muerte en los primeros años de vida. El tratamiento es quirúrgico, y se llama portoenterostomía de Kasai (KPE), siendo fundamental para intentar restablecer el flujo biliar hacia el intestino. El artículo tiene como objetivo analizar los principales aspectos de la AB como causa de ictericia obstructiva, abordando aspectos fisiopatológicos, anatomopatológicos, semiológicos, farmacológicos y médico-legales. El estudio se trata de una revisión bibliográfica exhaustiva basada en artículos seleccionados de las bases de datos PubMed, LILACS y SciELO, filtrados entre 2019 y 2024. Se utilizaron descriptores en tres idiomas, y se seleccionaron 15 artículos para componer el estudio, además de tres libros relevantes. El tratamiento estándar de la AB es la cirugía de Kasai, siendo más eficaz si se realiza antes de los 60 días de vida. Sin embargo, la tasa de fracaso es significativa, con muchos pacientes que eventualmente necesitan un trasplante de hígado. Factores como la edad del paciente en el momento de la cirugía y el restablecimiento del flujo biliar son críticos para el pronóstico, siendo así, es necesario que haya un diagnóstico precoz y que las intervenciones sean rápidas para que puedan mejorar la calidad de vida de los pacientes. Mientras la medicina no avanza en este campo, podemos sugerir el uso de guías que incluyan la carta de colores de heces y los signos/síntomas característicos y predecibles de la enfermedad, ya que no contamos con un tamizaje.
PALABRAS CLAVE: Colangiopancreatografia Retrógrada Endoscópica, Colangitis, Hepatomegalia, Hiperbilirrubinemia, Portoenterostomía Hepática.
ABSTRACT
Biliary atresia (BA) is a serious, idiopathic, and progressive condition, with a worldwide incidence of 1 in every 8,000 to 15,000 live births, being more common in Asian countries. The disease causes hyperbilirubinemia, acholic stools, dark urine, as well as hepatomegaly and splenomegaly, and if untreated, it can lead to death in the first years of life. The treatment is surgical and is called Kasai portoenterostomy (KPE), being essential to attempt to restore bile flow to the intestine. The article aims to analyze the main aspects of AB as a cause of obstructive jaundice, addressing pathophysiological, anatomopathological, semiological, pharmacological, and medico-legal aspects. The study is an exhaustive literature review based on articles selected from the PubMed, LILACS, and SciELO databases, filtered between 2019 and 2024. Descriptors in three languages were used, and 15 articles were selected to compose the study, in addition to three relevant books. The standard treatment for BA is the Kasai procedure, which is more effective if performed before 60 days of life. However, the failure rate is significant, with many patients eventually needing a liver transplant. Factors such as the patient’s age at the time of surgery and the restoration of bile flow are critical for prognosis; therefore, early diagnosis and prompt interventions are necessary to improve the patients’ quality of life. While medicine does not advance in this field, we can suggest the use of guides that include the stool color chart and the characteristic and predictable signs/symptoms of the disease, as we do not have a screening method.
KEYWORDS: Cholangitis, Endoscopic retrograde, cholangiopancreatography, Hepatomegaly, Hepatic portoenterostomy, Hyperbilirubinemia.
1. INTRODUCCIÓN
La atresia biliar (AB) es una colangiopatía obstructiva fibrosante progresiva que afecta tanto al sistema biliar extrahepático como al intrahepático, pero que, no obstante, se considera un proceso idiopático. Los signos clínicos son hiperbilirrubinemia directa o conjugada, heces acólicas, orina oscura, niveles variables de hepatoesplenomegalia e insuficiencia hepática progresiva. Si no reciben tratamiento, los recién nacidos afectados desarrollan una fibrosis que avanza rápidamente, lo que conduce a hipertensión portal y enfermedad hepática terminal, que progresa hasta la muerte en los primeros años de vida(1).
Esta enfermedad se presenta en todo el mundo y afecta 1 de cada 8.000 y 15.000 nacidos vivos, con mayor frecuencia en los países asiáticos. La mayoría de los artículos publicados demuestran un ligero predominio femenino, más comúnmente en niñas a término, con peso adecuado al nacer, que progresan con ictericia persistente entre la segunda y tercera semana de vida(1,2).
El diagnóstico precoz es fundamental para la portoenterostomía de Kasai (KPE), una intervención quirúrgica que elimina el árbol biliar extrahepático atrésico en un intento de restablecer el flujo de bilis al intestino mediante la creación de un conducto intestinal en Y (3). La AB es la principal causa de muerte neonatal, la causa más común de enfermedad hepática terminal en los primeros años y la indicación más frecuente de trasplante hepático pediátrico, representando entre el 40 y el 50% de los niños que reciben un trasplante hepático (4).
De esta manera, el problema central que busca abordar este trabajo es: ¿Cuáles son los principales aspectos fisiopatológicos, anatomopatológicos, semiológicos, farmacológicos y legales relacionados con la atresia biliar como causa de ictericia obstructiva, y cómo pueden contribuir estos conocimientos en el manejo de esta enfermedad? De esta manera, el objetivo principal de este artículo es, revisar y analizar los principales conceptos relacionados con la atresia biliar como causa de ictericia obstructiva. Para esto buscaremos: explorar los mecanismos fisiopatológicos implicados en la enfermedad, escribir los cambios anatómicos e histopatológicos observados, analizar los principales signos y síntomas para el diagnóstico clínico, evaluar las opciones farmacológicas disponibles y su efectividad, discutir los aspectos de la medicina legal y el impacto de las políticas sanitarias en la detección y el tratamiento de las enfermedades.
2. MATERIALES Y MÉTODOS
Se trata de un estudio bibliográfico, de tipo revisión de literatura, de carácter amplio que tiene como objetivo describir la atresia biliar como causa de ictericia obstructiva, desde un punto de vista teórico, a través del análisis e interpretación de la producción científica existente, cuyo enfoque es necesario conceptualizar las variantes involucradas.
Para realizar el estudio fueran utilizadas las siguientes bases de datos: PubMed (Biblioteca Nacional de Medicina), LILACS (Literatura Latinoamericana y del Caribe en Ciencias de la Salud) y SciELO (Scientific Electronic Library Online). Después de seleccionadas las plataformas, la pesquisa se llevó al cabo por intermedio de una búsqueda avanzada, utilizamos descriptores basados en la lista del DeCS y sus respectivos términos en inglés, portugués y español: “Atresia biliar” OR “biliary atresia” AND “icterícia obstrutiva”
OR “obstructive jaundice”. “Fisiopatologia” OR “pathophysiology” AND “atresia biliar”. “Anatomopatologia” OR “anatomopathology” AND “atresia biliar”, “Semiologia” OR “semiology” AND “atresia biliar”, “Farmacologia” OR “pharmacology” AND “atresia biliar”, “Medicina legal” OR “legal medicine” AND “atresia biliar”, “Gestão de saúde” OR “health management” AND “atresia biliar”.
Aplicamos filtros a los artículos obtenidos, incluidos los artículos publicados entre 2019-2024, escritos en inglés, portugués y español, dentro de los siguientes tipos: artículo clásico, ensayo clínico, artículo de revista, metaanálisis, revisión, revisión de integridad científica y revisiones sistémicas, al total encontramos 297 artículos.
Los criterios de exclusión fueron: artículos que no abordaron directamente la AB como causa de ictericia obstructiva, estudios duplicados en diferentes bases de datos y publicaciones que no presentaron claridad metodológica o que no aportaron datos relevantes para los objetivos de la revisión y artículos publicados con anterioridad al año de 2019. Los criterios de inclusión fueron: artículos publicados entre los años 2019 y 2024 con el objetivo de garantizar la actualidad de la información, publicaciones que abordasen la AB con énfasis en los siguientes temas: fisiopatología, anatomopatología, semiología, farmacología, medicina legal y gestión en salud.
Los artículos identificados en las bases de datos fueron inicialmente filtrados mediante la lectura de los títulos y resúmenes para comprobar su relevancia con la temática propuesta, seleccionándose 69 artículos. Luego de esta selección, se analizaron los textos completos para asegurar que cumplieran con los criterios de inclusión. De esta manera, al final sólo se seleccionó 15, entre los seleccionados se encuentran aquellos artículos considerados relevantes para la implementación del marco teórico que proponemos.
Tenemos como aporte, además de artículos, 3 libros para que el estudio bibliográfico se convierta en un instrumento indispensable para alcanzar los objetivos estipulados en este trabajo. Luego de toda la revisión, los datos extraídos de los artículos fueron organizados y presentados según la temática propuesta, al final utilizamos la aplicación Microsoft Word 2017 como herramienta para la construcción y finalización del trabajo.
3. MARCO TEÓRICO
La enfermedad en cuestión es una anomalía que afecta las vías biliares extrahepáticas e intrahepáticas de mayor tamaño y causa ictericia obstructiva en el primer mes de vida (5). Thomson designó la AB como una entidad independiente en 1892. En una revisión de un caso publicada en el Edinburgh Medical Journal, Holmes introdujo el concepto de enfermedades corregibles y no corregibles. Ladd describió el primer caso de atresia de las vías biliares corregible con cirugía en 1928 y Gross descubrió en 1953 que es la causa más común de ictericia obstructiva neonatal (6).
Después de años, la AB sigue siendo un proceso inflamatorio cuya causa desconocida provoca fibrosis y obliteración progresiva de la vía biliar extrahepática y lesiones del parénquima hepático y de la vía biliar intrahepática. Puede causar cirrosis precoz durante su desarrollo natural y causar la muerte antes de los tres años. Esta es la causa más frecuente de colestasis crónica infantil y la indicación más frecuente de trasplante de hígado. El diagnóstico precoz y el tratamiento quirúrgico en las primeras semanas de vida mejoran significativamente el pronóstico de la enfermedad(7).
Conforme la literatura consultada, podemos tener dos formas de AB: la primera, conocida como forma embrionaria o fetal, tiene un impacto entre el 10% al 35% de los casos. La segunda ocurre después del parto, entre el 65 y el 90% de los niños, denominada perinatal o no sindrómica. Se distingue entre una forma intrahepática o extrahepática que debe reclasificarse según la parte de las vías biliares afectadas, tal esquema anatómico fue una propuesta de la Asociación Japonesa de Cirujanos Pediatras (Figura 01) (6, 8).
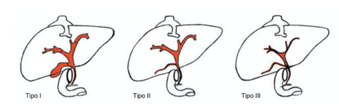
Figura 01. Tipos de atresia (8).
El tipo 1 (5-10%) es cuando el flujo biliar se obstruye a nivel del colédoco y la bilis suele estar en la vesícula biliar. El conducto biliar proximal puede ser quístico en estos casos. En el tipo 2, la obstrucción se encuentra al nivel del conducto hepático normal y la disección del porta hepático revelará conductos hepáticos diferentes, incluso con paredes gruesas y anormales. Esto es muy poco común en la mayoría de las series (1-2%). Por el contrario, el tipo 3 es más frecuente (>90%), presenta un alto nivel de obstrucción en el portal hepático y no hay conductos macroscópicos visibles porque el portal seccionado tiene un aspecto uniforme muy pálido(8).
En otras literaturas a parte de los tipos conocidos como forma embrionaria o fetal e la perinatal, hay otros dos tipos de AB: la quística que es representada por la presencia de una malformación quística cerca del sitio de la obstrucción del conducto biliar y la asociada a citomegalovirus (CMV), cuando los informes detectan una relación entre el drenaje biliar y una infección por CVM (6).
La incidencia global es significativamente variada, oscilando entre aproximadamente 1:5.000 y 10.000 nacidos vivos en Taiwán y Japón, y posiblemente en China, hasta aproximadamente 1:15.000 – 19.000 en Europa. Estas estimaciones suelen coincidir con la situación en América del Norte, donde los datos más recientes se basan en registros estadounidenses de 1:22.000. La heterogeneidad etiológica es una explicación obvia de esta variación, ya que la proporción de diferentes variantes cambia con el entorno local o con alguna predisposición genética (8). Cuando buscamos datos sobre estudios en Paraguay encontramos una ausencia de estudios nacionales.
La etiopatogenia tiene múltiples factores y ha sido objeto de estudios exhaustivos. Se han sugerido numerosos mecanismos patológicos potenciales, que pueden incluir infecciones virales, toxinas, variantes genéticas, anomalías inmunitarias o trastornos autoinmunes, microquimerismo materno, trastornos vasculares y defectos en el morfodesarrollo(9).
Cuando intentamos comprender la causa viral, la literatura sugiere que existe una lesión de conducción inicial causada por una respuesta inmune (CD4 a CD8 a Th1) a través de IL2, IL2, IL17, IFNγ, TNFα, que conduce a un proceso fibro obliterativo progresivo mediado por el sistema inmunológico ataca el epitelio biliar de los conductos biliares intra y extrahepáticos, lo que provocaría daño y destrucción de los conductos biliares(7).
Benjamin Landing sugirió por primera vez en 1974 que la infección viral del hígado y del árbol hepatobiliar causaba colangiopatías obstructivas como la AB. Varios virus, incluido el citomegalovirus (CMV), reovirus, virus del herpes humano, virus del papiloma humano, adenovirus, virus de Epstein Barr, virus de la hepatitis B, parvovirus B19 y rotavirus (RV), también conocido como virus colangiotrópicos, se han sugerido tener un papel potencial en este contexto(3).
Sobre la hipótesis de un defecto en la morfogénesis del tracto biliar, es atractiva la teoría de la patogénesis de la AB, especialmente si se tiene en cuenta la coexistencia de otras anomalías, como las anomalías de la simetría de los órganos viscerales que ocurren en el 10% al 20% de los lactantes con AB(9).
Genéticamente tenemos varios genes involucrados en el desarrollo anormal del sistema biliar y potencialmente en la patogénesis de la AB cuando abordamos la causa genética. En estudios de cohortes de niños con AB, encontraron variantes FOXA2 en miembros de una sola familia, así como variantes ADD3 y GPC1. PKD1L1 (que codifica una proteína ciliar) y EFEMP1 (que codifica la proteína de fibra elástica fibulina) son otras variantes reportadas en cohortes más grandes(9).
En otro estudio, hay una demostración de la conexión entre el alelo del antígeno leucocitario humano (HLA) B12 y los haplotipos HLAA9B5 y HLAA28B35 con la AB. También es posible que participen haplotipos como HLACw4/7, HLAA33, HLAB44 y HLADR6, así como polimorfismos específicos más comunes en algunos genes, como el SNP rs17095355 en 10q24 del gen ADD3(3).
Varias líneas de evidencia apuntan a una respuesta proinflamatoria que se dirige a los conductos biliares en pacientes con AB al relacionar los mecanismos proinflamatorios de la enfermedad. Según una teoría, un ataque viral o tóxico al epitelio biliar produce antígenos recién expresados en la superficie del epitelio de los conductos biliares. Estos antígenos son reconocidos por los linfocitos T en un entorno inmunológico genéticamente determinado, como la presencia de moléculas con mayor histocompatibilidad. Estos linfocitos T luego atacan las células inmunitarias causando daño(1).
Un modelo de rata con rotavirus ha demostrado la hipótesis predominante de que las citocinas proinflamatorias son esenciales para la patogénesis de la AB. Los estudios de agotamiento genético y celular en ratas muestran una variedad de eventos biológicos que causan obstrucción de los conductos biliares extrahepáticos, recapitulando algunas características de la enfermedad en humanos(1).
Los eventos comienzan con una infección viral que ataca el epitelio del conducto biliar y prepara macrófagos y células dendríticas (fase “inicial”). Esto sigue a la activación de las células NK que dañan los colangiocitos y alteran la continuidad de la mucosa (fase de lesión epitelial). Una amplificación de la respuesta inmune adaptativa por parte de las células T CD4 y CD8 y la liberación de citoquinas proinflamatorias forma un tapón celular en el sitio de la lesión epitelial (fase de obstrucción), seguido de la evolución hacia el depósito de colágeno para producir la atresia. fenotipo (Figura 2)(1).
En cuanto a la exposición a tóxicos ambientales, la única evidencia basada que respalda la agresión tóxica como factor causal de la AB es la agrupación temporal y espacial de los casos, que ocurrieron en 1964 y 1988 y produjeron brotes inusuales de lesión hepatobiliar en corderos y perros de Nueva Gales del Sur, Australia, con muestras patológicas que mostraron características similares a la patología observada en humanos(3).
En resumen, la etiología de la enfermedad es desconocida. Sin embargo, los estudios actuales muestran una compleja interacción entre la predisposición genética, los virus desencadenantes y la autoinmunidad progresiva que conduce al daño del tracto biliar, la fibrosis y la cirrosis biliar. Una mejor comprensión de los factores relacionados con la lesión epitelial de los conductos biliares servirá como base para futuras intervenciones terapéuticas destinadas a proteger el sistema biliar de futuras lesiones(9).
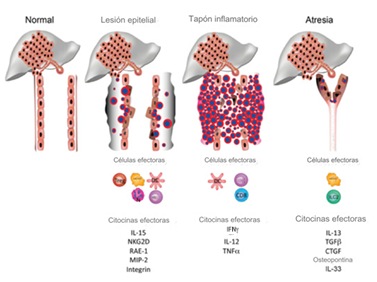
Figura 2. El modelo propuesto de patogénesis de la atresia biliar(1).
Para un diagnóstico e intervención oportunos, los signos y síntomas deben reconocerse temprano en cuanto a la presentación clínica. La ictericia persistente (evaluada por la escala de Kramer – Tabla 1), la acolia fecal y la hepatomegalia son los principales síntomas clínicos de la AB. Cuando se presentan estos síntomas en un recién nacido, es necesario sospechar de AB y llevar a cabo una evaluación diagnóstica completa. A partir de los 2 a 3 meses, aparecen signos de hipertensión portal como esplenomegalia y circulación colateral visible(8, 10).
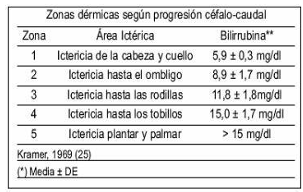
Tabla 1. Zonas Kramer y valor sérico de bilirrubina(8).
La pigmentación amarillenta de la piel y las mucosas causada por el depósito de bilirrubina en los tejidos se conoce como ictericia, que se manifiesta clínicamente cuando los niveles totales de bilirrubina superan los 2 mg/dl en lactantes y niños mayores. Los niños tienen concentraciones normales de bilirrubina sérica total inferiores a 1 mg/dl (bilirrubina directa inferior a 0,4 mg/dl)(11).
La hiperbilirrubinemia directa/conjugada o mixta (>1 mg/dl si Bb total <5 mg/dl o >20% si Bb total >5 mg/dl) puede ser secundaria a daño hepatocelular, alteración de la excreción canalicular, conducto biliar intra o extrahepático o un defecto de la redistribución enterohepática(11).
Los antecedentes familiares deben ser exhaustivos cuando se trata de anamnesis e incluir información sobre afecciones hepáticas, trastornos sanguíneos o anomalías cardíacas y vasculares. Utilizando las siguientes indicaciones como principales pistas de la historia clínica: la etapa inicial de la ictericia, coloración de heces y la tonalidad del orina, los antecedentes de colestasis intrahepática familiar progresiva (PFIC) o infecciones maternas previas, emisión de meconio retardado (FQ), uso de nutrición parenteral total (NPAC), historial de medicamentos y comida (intolerancia a la fructosa hereditaria) y historia de consanguinidad (EA, enfermedad metabólica genética/hereditaria) (12).
Además, los hallazgos de la exploración física deben incluir síntomas extrahepáticos como falta de actividad, somnolencia, características dismórficas, peso al nacer, retraso en el crecimiento y hallazgos dermatológicos, neurológicos, oftálmicos, cardíacos y hematológicos. Es importante cuestionar el color de las heces, ya que, si son totalmente acólicas, indican una obstrucción total del flujo de bilis, lo que puede indicar AB(12).
La atresia de las vías biliares requiere una combinación de pruebas clínicas, de laboratorio e imágenes para el diagnóstico. Los datos de laboratorio muestran un patrón con un aumento de la bilirrubina directa, gamma glutamiltransferasa (GGT) generalmente superior a 300 UI/L y un aumento de la fosfatasa alcalina. Se presenta con frecuencia junto con una leve elevación de las transaminasas (ALT/AST) y no está relacionada con insuficiencia hepática(7).
La ecografía es un examen importante para detectar la colestasis infantil, que con frecuencia se realiza como primer examen de detección. Este examen implica la visualización fallida de la vesícula biliar, también conocida como vesícula biliar hipoplásica fantasma, y la presencia de tejido hiperecoico triangular o tubular anterior a la vena porta. Los signos ecográficos de AB pueden incluir el “cordón triangular”, que representa el resto fibrótico de los conductos biliares atrésicos (Figura 3)(13).
La presencia de un cordón triangular en el examen ecográfico indica una tasa de precisión del 95% en el diagnóstico de atresia de las vías biliares extrahepáticas, una sensibilidad del 85% y una especificidad del 100%. Sin embargo, la colangiografía es necesaria para confirmar el diagnóstico, que puede realizarse mediante métodos invasivos o no invasivos(10).
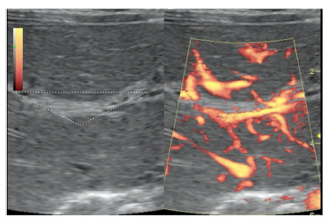
Figura 3: Las imágenes de ecografía Doppler en modo B y de potencia muestran un tejido fibroso ecogénico no vascularizado (borde triangular blanco) en el hilio hepático: signo del cordón triangular (13).
La colangiopancreatografía retrógrada endoscópica (CPRE) es un método válido, preciso y seguro para el diagnóstico cuando existe sospecha de AB, pero solo se puede realizar en centros especializados. Si persisten las deposiciones acólicas y se sospecha AB, se recomienda una CPRE y una biopsia hepática a más tardar dentro de las 4 a 6 semanas posteriores al nacimiento(12).
La CPRE puede ser útil para detectar la permeabilidad del árbol biliar extrahepático, con valores predictivos positivos y negativos altos para AB (sensibilidad 86% – 100%, especificidad 87% – 94%, valor predictivo positivo 88% – 96% y valor predictivo negativo 100%). Sin embargo, esta solicitud es para un endoscopista experto, un equipo de endoscopia pediátrica específico que no está disponible en muchos centros y un anestesista general, por lo que es poco común. La validez de otras técnicas, como la colangiorresonancia transhepática y la colecistocolangiografía transhepática percutánea, es insuficiente(12).
En un metaanálisis que compara varios métodos de diagnóstico, la biopsia hepática demostró ser más sensible, específica y precisa en el diagnóstico. Sin embargo, debido al riesgo inherente a su realización, solo se recomienda su uso en casos de dudas diagnósticas (10).
Cuando nos basamos en la histopatologia, encontramos las células inflamatorias y fibrosas que rodean los pequeños conductos biliares son probablemente restos del sistema original de conductos embrionarios. Se observan signos de colestasis y fibrosis en el parénquima hepático. Los neoconductillos biliares están proliferando. El drenaje biliar insuficiente puede llevar a una cirrosis terminal. En la progresión de la AB, el hígado muestra preservación de la arquitectura hepática básica, con proliferación de conductos biliares, estasis biliar canalicular y celular, conductos portales o edema perilobulillar y fibrosis (Figura 4) (1).
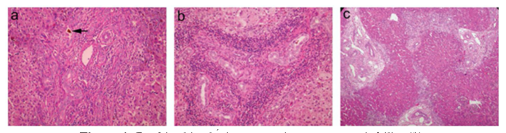
Figura 4. Cambios histológicos en pacientes con atresia biliar (1).
Todavía, no hay un tratamiento específico para la AB en la actualidad, puesto esto, el tratamiento implica en una intervención quirúrgica que facilita la liberación adecuada de bilis al intestino, lo que reduce el riesgo de fibrosis hepática e infiltrados inflamatorios. La hepatoportoenterostomía de Kasai es el primer método de selección en niños, la anastomosis en Y de Roux del yeyuno con los conductos biliares garantiza la salida de bilis y secreción pancreática(6).
Desde hace más de sesenta años, esta cirugía se ha convertido en un procedimiento quirúrgico estándar. En el 80% de los casos, la liberación de bilis se logra si la operación se realiza antes del día 60 de vida. La progresión hacia la cirrosis y la hipertensión portal es posible incluso después de una intervención quirúrgica exitosa. La tasa de supervivencia es del 25% al 30%(8).
Tras la cirugía, un 30% no restablecerá el flujo de bilis y otro 30% lo hará parcialmente, requiriendo ambos grupos de trasplantes de hígado en los siguientes meses. Se estima que, del 40% restante en el que se restablece el flujo de bilis, alrededor del 70% necesitará un trasplante de larga duración. Esto, en su conjunto, implica una necesidad de trasplante para estos pacientes en algún momento de su infancia en torno al 70-80% de los casos, pero también, dados los buenos resultados del trasplante, una supervivencia global del 90% a los 10 años de vida(7). Pero hay casos que hay indicaciones de un trasplante de hígado tras una portoenterostomía que son: falta de drenaje biliar, signos de retraso en el desarrollo o sus secuelas y presencia de complicaciones/efectos secundarios socialmente inaceptables.
Al citar el pronóstico, tenemos dos tipos de factores, los modificables (edad en la cirugía, recuperación del flujo de bilis después de una cirrosis y la experiencia del equipo quirúrgico) y no modificables (la atresia sindrómica, la forma anatómica completa e infección por CMV (7). Con respecto al diagnóstico diferencial, es muy amplio y complejo, pero los principales incluyen hepatopatía relacionada con la nutrición parenteral total (TPN), quiste colédoco, síndrome de Alagille, deficiencia de antitripsina, proteína de resistencia a múltiples fármacos 3 deficiencia de MDR3, fibrosis quística, colangitis esclerosante neonatal (NSC) o cualquier otra causa de obstrucción mecánica. (3).
Al relacionar los casos que llegan a la autopsia, tenemos que, la cirugía (figura 5), la colangitis y la septicemia son las causas de muerte más comunes. El 50% a 90% de los casos desarrollan colangitis, generalmente dentro de los primeros tres meses, pero generalmente se debe a bacterias gramnegativas. También puede ocurrir años después de la cirugía y empeorar el pronóstico(14).
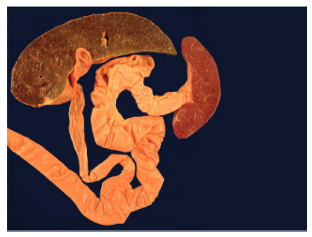
Figura 5: autopsia, portoenteroanastomosis en Y de Kasai; El hígado es de tamaño reducido con una superficie nodular de aspecto fibroso, también se observa esplenomegalia congestiva (14).
4. RESULTADOS Y DISCUSIÓN
La AB es una colangiopatía obstructiva que afecta los conductos biliares, es una enfermedad hepática cuya etiología y patogénesis no se comprenden completamente. Según Bezerra et al., dicha enfermedad se trata de una reacción inflamatoria exacerbada de los conductos biliares en respuesta a la infección por ciertos tipos de virus, en individuos con predisposición genética a la desregulación en la producción de citocinas y otras sustancias proinflamatorias. Porth (2019), describe que la AB se presenta en el período neonatal con ictericia persistente, orina oscura, heces pálidas y hepatomegalia (15). Es fatal cuando no tratada, en estos casos con una supervivencia reportada de menos del 10% a los 3 años de edad.
A lo largo de este estudio se observó que el tratamiento definitivo para la atresia de las vías biliares es quirúrgico, hecho mediante portoenterostomía de Kasai, con el objetivo de restaurar el flujo biliar y prevenir la rápida progresión del daño hepático. Según Schreiber et al., en los países desarrollados, la edad en el momento de la cirugía es aproximadamente 60 días y este hecho asociado a la experiencia de los centros de referencia en la realización del procedimiento, ha sido el factor más importante para mejorar resultados de acuerdo con la literatura(16).
Aunque todos los datos apuntan hacia una estrategia de portoenterostomía de Kasai de “cuanto antes, mejor”, Serpa et al. en su estudio demostra que los niños con AB suelen acudir al gastroenterólogo pediátrico y/o al cirujano pediátrico a una edad “tardía” y que la vigilancia de la presencia de heces pálidas por parte de los profesionales sanitarios o los padres no forma parte de los cuidados habituales del niño sano (8), dato que nos prende un alerta rojo, pues cuidados sencillos pondrian mejorar la sospecha y consecuentemente el diagnostico.
En el estudio de Dmytriieva et al., describe que en China y Taiwán, se añadió al historial médico del recién nacido una tarjeta de prueba con siete colores diferentes de heces, tres de los cuales eran anormales, así, durante el primer mes de vida, los padres del niño compararon el color de las heces del niño con la tarjeta de prueba adjunta y luego la enviaron a los centros de detección, con eso 75% de los casos de AB fueron diagnosticados y tratados precozmente, lo que redujo la mortalidad y mejoró la calidad de vida de estos pacientes (17).
La literatura de la última década muestra una calidad de vida (CV) significativamente inferior en los niños con AB en comparación con sus compañeros sanos, con un gran tamaño del efecto en la CV física y un tamaño del efecto de moderado a grande en la CV psicosocial (18). De esta manera, Rodijk et al. manifiestan que la CV física fue significativamente menor en comparación con pacientes sanos.
De manera semejante, Liang et al. exponen que, tras la cirugía de Kasai, los niños experimentaron salud física, función emocional, función social, función cognitiva y calidad de vida significativamente inferiores frente a niños sanos(19). De esta manera, debemos tener en cuenta los hallazgos clínicos para ayudar en el diagnóstico precoz.
5 CONSIDERACIONES FINALES
La AB es una enfermedad compleja de etiología multifactorial que se manifiesta como hiperbilirrubinemia conjugada en neonatos y lactantes. La AB tiene consecuencias muy perjudiciales para la salud del lactante, con una rápida progresión a hipertensión portal y enfermedad hepática terminal si no se trata de manera oportuna.
Los niños con sospecha de AB necesitan un examen morfológico del núcleo de tejido de una biopsia hepática percutánea. Los predictores histológicos más fuertes de la obstrucción de los conductos grandes son los tapones biliares ductulares y el edema del estroma portal.
La portoenterostomía de Kasai sigue siendo el procedimiento habitual para tratar la atresia de las vías biliares en la actualidad, dejando atrás los procedimientos invasivos totalmente abiertos para el uso de técnicas menos invasivas por laparoscopia. Si se realiza en los primeros tres meses de vida, tiene mejor pronóstico, pero es un procedimiento que fracasa en un número significativo de casos.
El tratamiento con este procedimiento es eficaz en el restablecimiento del flujo biliar y previene complicaciones como la progresión a la cirrosis biliar. Sin embargo, es importante recordar que, debido a la progresión de la enfermedad, se pueden presentar complicaciones como colangitis e hipertensión portal. El diagnóstico y tratamiento temprano, como en cualquier otro tipo de patología, puede evitar complicaciones y muertes tempranas y asegurar una calidad de vida para el paciente.
Es esencial mantener los niveles de bilirrubina adecuados después de la cirugía, por lo que es esencial monitorear al paciente. Esto es uno de los factores predictivos de la supervivencia de los pacientes. Lamentablemente, la mayoría de los pacientes con esta enfermedad desarrollan una enfermedad hepática terminal y requieren un trasplante de hígado, lo que conduce a una mayor morbimortalidad.
Es evidente que se requiere una comprensión urgente de la fisiopatología de la AB para desarrollar una cura primaria y obtener un mejor pronóstico de la enfermedad en los niños afectados. En espera de los avances médicos en la AB, podemos sugerir la implementación por parte de las maternidades e incluso en la Atención Primaria de Salud, el uso de guías/tarjetas que incluyan la carta de colores de heces como un cribado para la AB, bien como los signos/síntomas característicos de la enfermedad para que los padres puedan estar atentos, ya que no contamos con un tamizaje para AB.
6. REFERÊNCIAS
1. Bezerra JA, Asai A, Tiao G, Mullapudi B, Balistreri WF. Biliary atresia and other disorders of the extrahepatic bile ducts. In: Liver Disease in Children. Cambridge University Press; 2021 [citado el 30 de septiembre de 2024]; p. 162–81. Disponible en: https://doi.org/10.1017/9781108918978.011
2. Martínez RV, Osoria AJP, Guirado AR, Duvergel AP, Berriz DB, García CS, et al. Guía de práctica clínica en atresia de las vías biliares. Revista Cubana de Pediatría [Internet]. 2020 [citado el 30 de septiembre de 2024]; 92(4). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312020000400016
3. Vij M, Rela M. Biliary atresia: pathology, etiology and pathogenesis. Future Sci OA [Internet]. 2020 [citado el 30 de septiembre de 2024]; 6(5). Disponible en: http://dx.doi.org/10.2144/fsoa-2019-0153
4. Feldman AG, Sokol RJ. Neonatal cholestasis: Updates on diagnostics, therapeutics, and prevention. Neoreviews [Internet]. 2021 [citado el 30 de septiembre de 2024]; 22(12):e819–36. Disponible en: http://dx.doi.org/10.1542/neo.22-12-e819
5. Borstnar CR, Cardellach F, editores. Farreras Rozman. Medicina Interna. 19a ed. Elsevier; 2020.
6. Paredes SAH, Auz SAM, Bombón EGC, Ortega MBD. Cirugía de Kasai. RECIMUNDO [Internet]. 2021 [citado el 30 de septiembre de 2024]; 5(1):138–45 Disponible en: http://dx.doi.org/10.26820/recimundo/5.(esp.1).nov.2021.138-145
7. Tomé LF, Remacha EF. Colestasis en el lactante. Aeped.es. [Internet]. 2023 [citado el 30 de septiembre de 2024]; Disponible en: https://www.aeped.es/sites/default/files/documentos/28_colestasis.pdf
8. Sarmiento APS, Merchán JPP. Atresia de las vías biliares, reporte de caso y revisión de la literatura: Atresia of the bile ducts, case report and review of the literature. LATAM Revista Latinoamericana de Ciencias Sociales y Humanidades [Internet]. 2023 [citado el 30 de septiembre de 2024]; 4(1). Disponible en: http://dx.doi.org/10.56712/latam.v4i1.383
9. Holcomb GW, Murphy JP, Peter SD, editores. Holcomb Y Ashcraft. Cirugia Pediatrica. 7a ed. Elsevier; 2021.
10. Pinto BB, Salvador IIT. Desentrañar la atresia de las vías biliares: una revisión exhaustiva. Revista Contemporánea [Internet]. 2023 [citado el 30 de septiembre de 2024]; 3(8):11553–64. Disponible en: http://dx.doi.org/10.56083/rcv3n8-088
11. Uhalte AS, García CP, Torres IA. Diagnóstico diferencial de la ictericia en el lactante y niño mayor. Fapap.es. [Internet]. 2022 [citado el 30 de septiembre de 2024]; 15(4):138-44. Disponible en: https://fapap.es/files/639-2126-RUTA/01_Puesta_al_dia_en_Ictericia_v.pdf
12. Nicole J, Lemos M. Protocol of Investigation of Neonatal Cholestasis ¿Cuál es el abordaje diagnóstico de un recién nacido con Colestasis Neonatal? – Una revisión bibliográfica [Internet]. 2021 [citado el 30 de septiembre de 2024]; Disponible en: https://ubibliorum.ubi.pt/bitstream/10400.6/11401/1/8258_17673.pdf
13. Di Serafino M, Gioioso M, Severino R, Esposito F, Vezzali N, Ferro F, et al. Ultrasound findings in paediatric cholestasis: how to image the patient and what to look for. J Ultrasound [Internet]. 2020 [citado el 30 de septiembre de 2024]; 23(1):1–12. Disponible en: http://dx.doi.org/10.1007/s40477-019-00362-9
14. Sanz CR, Delgado CTC. Causas de muerte en niños con tratamiento quirúrgico de atresia de vías biliares. Estudio de autopsias. Acta Pediátrica México [Internet]. 2022 [citado el 30 de septiembre de 2024]; 43(3):147. Disponible en: http://dx.doi.org/10.18233/apm43no3pp147-1552341
15. Norris TL, Lalchandani R. Porth. Fisiopatologia. 10a ed. Baltimore, MD, Estados Unidos de América: Wolters Kluwer Health; 2019.
16. Schreiber RA, Harpavat S, Hulscher JBF, Wildhaber BE. Biliary atresia in 2021: Epidemiology, screening and public policy. J Clínica Med [Internet]. 2022 [citado el 01 de octubre de 2024]; 11(4):999. Disponible en: http://dx.doi.org/10.3390/jcm11040999
17. Dmytriieva K, Dmytriiev K, Vidiscak M, Vidiscak V. Diagnosis and treatment of biliary atresia in children. Pain Med [Internet]. 2022 [citado el 01 de octubre de 2024]; 7(1):40–6. Disponible en: http://dx.doi.org/10.31636/pmjua.v7i1.4
18. Rodijk L. Biliary atresia: neurodevelopment and quality of life. University of Groningen Press; 2020 [citado el 01 de octubre de 2024]; Disponible en: https://doi.org/10.33612/diss.133865199
19. Liang Y, Yu H, Shu F, Huang W, Jiang X, Xu Z, Zhang T, Xiang B, Jin S. Factors influencing the quality of life in children after biliary atresia treatment. Transl Pediatric [Internet]. 2021 [citado el 01 de octubre de 2024]; 10(10):2496-505. Disponível em: https://doi.org/10.21037/tp-21-391
1 Discente del 6to período de Medicina. ORCID: 0009-0003-2782-3111
2 Discente del 6to período de Medicina. ORCID: 0009-0006-1885-7493
3 Discente del 6to período de Medicina. ORCID: 0000-0003-2668-6963
4 Discente del 6to período de Medicina. ORCID: 0009-0009-4092-8480
5 Discente del 6to período de Medicina. ORCID: 0009-0007-4088-6888
6 Discente del 6to período de Medicina. ORCID: 0009-0002-0017-867X
7 Discente del 6to período de Medicina. ORCID: 0009-0008-5467-5691
8 Discente del 6to período de Medicina. ORCID: 0000-0002-2699-4475
9 Discente del 6to período de Medicina. ORCID: 0005-8342-4953
10 Discente del 6to período de Medicina. ORCID: 0009-0002-1673-6461