RISK ANALYSIS IN THE PROJECT MANAGEMENT OF THE IMPLEMENTATION OF THE ASEPTIC PROCESS IN THE INJECTION POWDER FILLING AREA – MEDIA FILL
REGISTRO DOI: 10.5281/zenodo.10080840
Edimar Telécio¹
Poliana Triers¹
Rosa Silva Lima2
Bruna de Oliveira3
RESUMO
Introdução: Aanálise de risco na gestão do projeto de implantação do processo asséptico na área de envase de pós injetáveis, com destaque para o teste de Media Fill, desempenha um papel fundamental na garantia da qualidade, segurança e conformidade dos produtos farmacêuticos estéreis. Objetivo: Este estudo teve como objetivo realizar uma análise de risco na gestão de projeto da implantação do processo asséptico na área de envase de pós injetáveis, com foco no teste de Media Fill. Metodologia: Este estudo consiste em uma análise de risco, aplicando a ferramenta de gestão FMEA (Failure Mode and Effect Analysis), com o objetivo de gerenciar os principais riscos observados na implementação do processo asséptico na área de envase. Para a análise de risco, utilizaram-se dados hipotéticos. Os principais dados foram apresentados em uma tabela FMEA, seguida das avaliações necessárias para o estudo. Resultados: A análise de risco desempenha um papel crucial nesse contexto, permitindo a identificação e avaliação dos riscos potenciais relacionados à contaminação do ambiente asséptico e ao processo de envase. Para isso, são empregadas diversas técnicas, como brainstorming, análise SWOT e análise de causa e efeito, a fim de mapear os riscos e suas possíveis causas. A seleção e qualificação dos equipamentos são aspectos críticos na implantação do processo asséptico. É essencial escolher materiais de construção adequados e realizar a validação da limpeza, esterilização e integridade dos sistemas de filtragem. Além disso, a manutenção regular e o treinamento técnico adequado são fundamentais para garantir a eficácia dos equipamentos ao longo do tempo. A importância do Media Fill reside na sua capacidade de simular condições reais de produção, permitindo a detecção de potenciais riscos de contaminação antes da produção em escala real.
Palavras-Chave: Análise de risco; Processo asséptico; Media Fill.
ABSTRACT
Introduction: Risk analysis in the management of the project for implementing the aseptic process in the filling of injectable powders, with a focus on Media Fill testing, plays a fundamental role in ensuring the quality, safety, and compliance of sterile pharmaceutical products. Objective: This study aimed to conduct a risk analysis in the project management of the implementation of the aseptic process in the filling of injectable powders, with a focus on Media Fill testing. Methodology: This study consists of a risk analysis, applying the FMEA (Failure Mode and Effects Analysis) management tool, with the objective of managing the main risks observed in the implementation of the aseptic process in the filling area. For the risk analysis, hypothetical data were used. The main data were presented in an FMEA table, followed by the necessary evaluations for the study. Results: Risk analysis plays a crucial role in this context, enabling the identification and assessment of potential risks related to contamination of the aseptic environment and the filling process. Various techniques, such as brainstorming, SWOT analysis, and cause and effect analysis, are employed to map the risks and their possible causes. Equipment selection and qualification are critical aspects in the implementation of the aseptic process. It is essential to choose suitable construction materials and validate the cleanliness, sterilization, and integrity of the filtration systems. Furthermore, regular maintenance and appropriate technical training are fundamental to ensuring the effectiveness of the equipment over time. The importance of Media Fill in its ability to simulate real production conditions, allowing for the detection of potential contamination risks before actual scale production.
Keywords: Risk analysis. Aseptic process. Media Fill.
INTRODUÇÃO
As estratégias de gerenciamento de riscos são as táticas utilizadas para lidar com eles e entender suas possíveis consequências. Essas estratégias devem ser incluídas em um plano de gerenciamento de riscos, que é um processo documentado de como sua empresa ou equipe identificará e abordará os riscos emergentes (MARQUES JUNIOR, 2000).
A análise dos elementos do processo de gestão de risco permite gerar uma ferramenta metodológica, em especial em indústrias que atuam com processos químicos, como é o caso da indústria farmacêutica, que precisa ter um controle rigoroso de suas operações, visto que se trata uma área que precisa de um elevado grau de precisão, assepsia, entre outros fatores para o funcionamento da empresa (VARGAS, 2010).
Nesse contexto, durante a fabricação de medicamentos, muitos produtos e seus ingredientes ativos podem ser contaminados por outros produtos farmacêuticos e ingredientes ativos, agentes de limpeza, microorganismos ou outros materiais, como lubrificantes, partículas transportadas pelo ar, matérias-primas, intermediários e auxiliares (COX et al., 2016). Cox et al., (2016) destacam ainda que em muitos casos, o mesmo equipamento pode ser usado para a produção de diferentes produtos subsequentes.
Dentro da gestão de risco, neste caso voltado para a indústria farmacêutica, tem-se a área de validação de processo. Os estudos de validação são uma parte essencial e são realizados de acordo com um plano mestre de validação. Entre os processos a serem validados estão os procedimentos de limpeza dos equipamentos de fabricação de medicamentos. Para realizar a validação de limpeza, é necessário preparar um protocolo que descreva as diretrizes e a sequência dos passos a serem seguidos (AGALLOCO; AKERS, 2021).
A validação do processo de envase asséptico ou “Media Fill Test” é a simulação de um processo de fabricação e/ou dosagem asséptico, no qual o produto é substituído por meio de cultura que promove o crescimento microbiano (WHITE, 2010).
Um teste chamado de Media Fill (também conhecido como simulação de processo) é um teste microbiológico fundamental que é realizado para avaliar o desempenho de um procedimento de fabricação asséptico, substituindo o medicamento ou bebida por um meio de cultura estéril (MCIVER, 2019).
A fabricação asséptica é um processo complexo usado nas indústrias farmacêutica. Dessa forma, As Boas Práticas de Fabricação (BPF) exigem que as empresas farmacêuticas e de bebidas realizem regularmente testes de preenchimento de mídia para verificar o status microbiológico de seu processo de produção asséptico (MCIVER, 2019).
Este trabalho teve como objetivo descrever a importância do gerenciamento do risco na execução do projeto da implantação do processo asséptico na área de envase de pós injetáveis (Media Fill).
Gerenciamento de risco na gestão de projetos
A globalização levou as empresas a serem forçadas a competirem por baixo custo em todo o mundo. Isso leva a uma alta demanda por crescimento e inovação, com isso os projetos passaram a ter uma importância cada vez maior nas empresas. Os projetos são iniciativas únicas, como lançar novos produtos, novas organizações ou novos empreendimentos, ou ainda, melhorar produtos existentes e investir na infraestrutura da empresa. Os projetos são os elementos executores de mudanças nas organizações que permitem às organizações sobreviver e crescer (CLELAND, 1994; SHENHAR & DVIR, 2007).
Entretanto, verifica-se cada vez mais um elevado número de problemas e fracassos nas realizações desses projetos. Muitos desses problemas poderiam ser previstos no processo de gestão de risco do projeto, com isso riscos são circunstâncias ou acontecimentos inesperados que afetam o projeto de alguma forma. Apesar da importância dos projetos nas organizações, a maioria dos projetos não cumpre suas metas iniciais de prazo, custo, qualidade e negócios (MARQUES JUNIOR, 2000).
São diversas as causas que levam ao não cumprimento das metas dos projetos. São apontadas por diversos autores: fatores ligados ao planejamento, falta de definição do que seja sucesso, falta de apoio da alta administração, falta de métricas para controle e falta de alinhamento dos interessados com critérios de sucesso, a falta de alinhamento com o negócio e a falta de envolvimento da alta administração (MARQUES JUNIOR, 2000; HARTMAN & ASHRAFI, 2002; FICHTER, 2003).
Devido essa baixa taxa de sucesso nos projetos, tem se observado questionamentos na gestão dos projetos nas empresas. Williams (2005) e Maylor (2002) identificaram a abordagem tradicional que tem como foco o desenvolvimento de técnicas e ferramentas de planejamento e controle que racionalizam e normatizam a gestão dos projetos. Contudo, a utilização destas técnicas leva a um cenário estável e previsível, porém esse ambiente não é observado no cenário atual, que é caracterizado por instabilidade e imprevisibilidade, devido a utilização de grandes tecnologias no mercado atual. Portanto tais fatos tem gerado críticas a essa abordagem tradicional na gestão de projetos. Onde é sugerido uma a utilização de ferramentas sofisticadas para gestão de projetos e a utilização de estudos com abordagens adaptativas na gestão de projetos que busquem a praticas adaptativas e critérios de sucesso (PINTO; SLEVIN, 1988; MORRIS, 1994; MEYER,2002; MILLS et al., 2002; MIDLER, 2002; MURIITHI; CRAWFORD, 2003; JAAFARI, 2003; WESTERVELD, 2003; WILLIAMS, 2005; CICMIL, 2006; HALLGREEN; MAANINEN-OLSSON, 2005; LEYBOURNE, 2007; SHENHAR; DVIR, 2007; DONK; MOLLOY, 2008 HARTMAN, 2008).
Segundo Ricardo Vargas, os pontos chaves para o sucesso na gestão de projetos é com base no conhecimento sobre cenário organizacional e os fatores internos e externos da empresa, levando em consideração os elementos próprios do projeto, como a gerência de escopo, a gerência na análise de riscos, o gerenciamento de recursos humanos e deve ter um gerente de projeto que possua habilidades de liderança, comunicação, senso de justiça, ser claro, possuir uma boa reputação e principalmente uma formação que qualifique o gerente a lidar com todos esses conhecimentos e recursos que envolvem a Gerência de Projetos (VARGAS, 2010).
O gerenciamento de risco deve ser iterativo e auxiliar as organizações no estabelecimento de estratégias para alcançar os objetivos e dar subsídios na tomada de decisões na empresa. O gerenciamento de risco baseia-se nos princípios, estrutura e processos delineados conforme ilustrado na Figura 1. Estes componentes podem já existir total ou parcialmente na organização, porém podem ser adaptados ou melhorados, para um gerenciamento de risco eficiente e eficaz (ABNT, 2018; PMBOK, 2017).
Figura 01. Princípio, estrutura e processo.
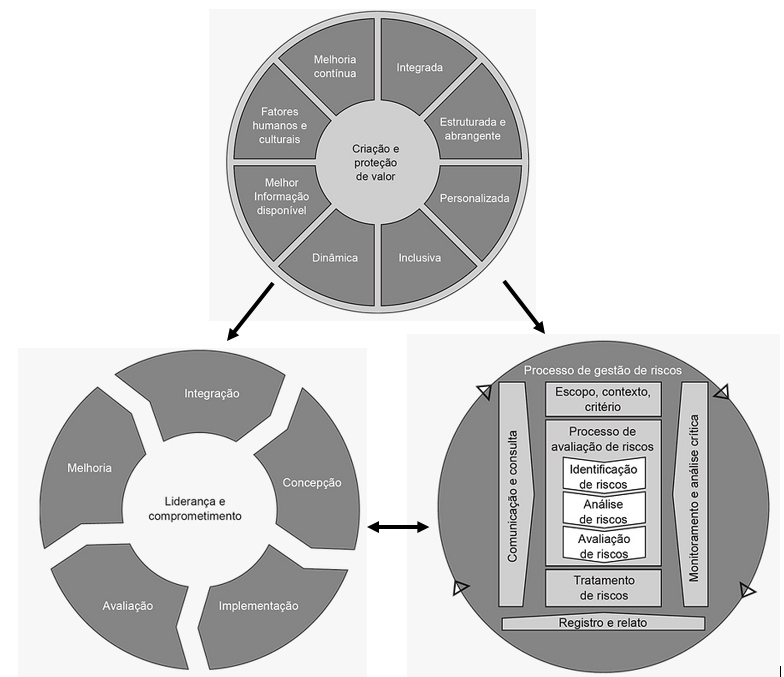
Fonte: ABNT, 2018.
ANÁLISE DE RISCO E FERRAMENTAS DE ANÁLISE DE RISCO
A gestão do risco como já citado é fundamental para o bom gerenciamento de projetos na Indústria Farmacêutica. Para tal é importante que se utilize as ferramentas de análise de risco correto para o seu projeto. A indústria farmacêutica segue normas de padrão de qualidade como a RDC 658/2022 que regulamenta as Boas Práticas de Fabricação, a utilização destas ferramentas corroboram para que o produto final tenha a qualidade, eficácia e segurança necessária para o paciente.
A avaliação dos riscos é o resultado da identificação, análise e classificação dos riscos associados à exposição aos perigos. Uma descrição bem definida de uma condição de risco ou perigo é determinante para um bom início da avaliação destes, facilitando inclusive na identificação da melhor ferramenta a ser empregada.
O processo de avaliação dos riscos é composto por três etapas necessárias para a conclusão desta fase.
A primeira etapa a ser realizada é a identificação dos riscos. Nesta etapa é realizada a busca das informações relativas ao perigo a que o processo ou produto está sujeito. Compromete-se basicamente a responder o questionamento do que pode ocorrer de errado, identificando as possíveis consequências desta questão, com base no conhecimento gerado pelas informações obtidas dos dados históricos, análises teóricas, opiniões técnicas e repostas a instruções ou perguntas (ICH Q9, 2005; ABNT, 2012; ABNT, 2018).
A etapa posterior é a análise dos riscos identificados e o processo qualitativo ou quantitativo pelo qual é possível se obter a vinculação entre a probabilidade e a severidade de um determinado dano. É a estimativa da correlação entre o risco e o perigo identificado. Em algumas ferramentas de gestão de risco é possível considerar a detecção do dano, desta forma este critério também estará correlacionado na estimativa do risco (ICH Q9, 2005).
A análise dos riscos pode ser conduzida pelo emprego de ferramentas com metodologia quantitativa, qualitativa ou semiquantitativa. A opção por qual metodologia a ser empregada dependerá do volume de informações que se possui sobre o processo, produto ou sistema (ABNT, 2012; ABNT, 2018).
Finalizadas as etapas de identificação e análise dos riscos é possível iniciar sua classificação em determinados níveis. Nesta etapa é possível comparar os valores obtidos pela análise de riscos com os critérios previamente estabelecidos (ICH Q9, 2005; ABNT, 2012; ABNT, 2018).
Um grande aliado nos estudos dos riscos de um projeto são as ferramentas de gestão de risco. Dentre as ferramentas de gestão de risco algumas se destacam para determinadas áreas, para o uso pretendido destaca-se a FMEA ou análise de modo de falha e efeito, que é uma metodologia sistemática que permite identificar falhas potenciais de um sistema, projeto e/ou processo com objetivo de minimizar ou eliminar os riscos/falhas antes que de produzir um produto. Com a sua utilização diminuem-se as chances de o produto ou processo falhar, aumentando a confiabilidade (CEDGEI, 2008; LOBO, 2020).
A FMEA foi desenvolvida com enfoque no projeto de novos produtos e processos, por sua utilidade, passou a ser aplicada de diversas maneiras para diminuir falhas de produtos, processos e para reduzir os desvios em processos administrativos (LOBO, 2020).
Para um melhor aproveitamento da aplicação da ferramenta FMEA (Failure Mode and Effect Analysis), um líder deve ser nomeado e este deve montar uma equipe de profissionais multidisciplinar, preferencialmente com especialistas de diferentes áreas envolvidas na proposta da avaliação. Desta forma o time de especialistas poderá definir os modos de falha de cada componente, identificando os efeitos e propondo medidas de ação que deverem ser tomadas, dentro de um consenso. No caso de estar sendo usada a metodologia que possibilite a quantificação, este também será responsável por avaliar a severidade, probabilidade ou frequência de ocorrência e detectabilidade do sistema (ICH, 2005; CEDGEI, 2008; LOBO, 2020).
PROCESSO ASSÉPTICO NA ÁREA DE ENVASE DE PÓS INJETÁVEIS (MEDIA FILL)
Na produção de medicamentos altamente ativos ou altamente sensibilizantes, as instalações devem utilizar sistemas adequados de tratamento do ar, com sistema de exaustão adequado, com baterias de filtro capazes de impedir que os resíduos do produto sejam libertados para o ambiente. Para a produção de um medicamento injetável, que deve ser estéril, o sistema de exaustão deve ser organizado mantendo um fluxo laminar que mantenha pressões diferentes em cada sala envolvida no processo, removendo partículas do interior das salas. Além disso, é importante controlar as manutenções do sistema de exaustão, como troca de filtros, para que estas atividades também sejam seguras (LEONARDI, 2010).
Os processos devem ser avaliados desde o início do projeto farmacêutico, avaliando os fluxos de matérias-primas, intermediários, produtos a granel, produtos semiacabados, produtos acabados, materiais de embalagem, reagentes, outros tipos de materiais, vários tipos de resíduos, e pessoas, tendo em conta cada tipo de paramentação e ponto de troca de coletes. Ademais, as áreas dentro do setor de produção devem ser concebidas para controlar a geração, disseminação e monitorar o número de partículas viáveis e não viáveis, a fim de evitar a contaminação cruzada e reduzir o risco de qualquer etapa de fabricação ou controle ser omitida ou mal aplicada. Para facilitar a limpeza, devem ser desenvolvidas e instaladas tubagens, acessórios, pontos de ventilação, e outras instalações. A iluminação nas áreas de produção, especialmente onde são manuseados controles visuais, deve ser adequada. (ANVISA, 2013; BRASIL, 2022).
Os Sistemas de Aquecimento, Ventilação e Ar-Condicionado (HVAC) devem ser instalados, avaliados e monitorados em áreas utilizadas na fabricação de produtos esterilizados, de acordo com as diretrizes técnicas pré-determinadas pelas organizações internacionais (OMS, ISO e outras), bem como pela Associação Brasileira de Normas Técnicas (ABNT) (BRASIL,2022).
As salas de produção nas fábricas de medicamentos devem ser limpas e classificadas em diferentes níveis. As quais são classificadas nos graus A, B, C, e D em termos das boas práticas de fabricação (BRASIL, 2022).
Todos os processos devem ser validados antes de iniciar a produção dos medicamentos.
A validação dos processos assépticos na produção de medicamentos deve incluir uma simulação utilizando meios de cultura conhecido como teste de enchimento simulado ou Media Fill. Os testes simulam operações assépticas de envase, substituindo o produto por um meio de cultura previamente estéril, durante toda a linha do processo de envase, para garantir que os métodos utilizados possam produzir produtos estéreis. Os recipientes não devem apresentar crescimento microbiano ou exceder um limite pré-definido para passarem neste teste (IN35/2019).
Segundo o relatório técnico da associação farmacêutica norte-americana, PDA, de 1996, o objetivo do método Media Fill é “demonstrar a capacidade do processo de manusear produtos de forma estéril e qualificar ou certificar os operadores da área asséptica para cumprir com os requisitos das boas práticas de fabricação” (BRASIL, 1996).
O teste é realizado em todas as condições rotineiras, utilizando os mesmos maquinários, processos, recipientes primários e todos os elementos diretamente relacionados ao processo feito na rotina da produção. Durante o envase, o processo todo deve ser desafiado, contemplando intervenções que normalmente acontecem, sendo elas, mecânicas, queda de energia, troca de filtro estéril, ajuste de máquina por colaborador, troca de turno, saída de colaborador para refeição, entre outros. Após o envase, os recipientes primários são acondicionados em caixas ou bandejas e devidamente identificados. Todas as unidades devem ser inspecionadas visualmente, para verificar a existência de fissuras, defeitos de fechamentos e/ou recipientes vazios, para serem descartados, caso necessário (BRASIL, 2010).
As unidades envasadas são incubadas a uma temperatura de 20°C a 25°C, por um período mínimo de 7 dias, na posição vertical e com a tampa para cima. Em seguida, são mantidas em incubação a uma temperatura de 30°C a 35°C, por mais um período mínimo de 7 dias, na posição contrária, totalizando o tempo mínimo de incubação de 14 dias em temperatura controlada. As unidades são inspecionadas nos intervalos de 7 e 14 dias e, se houver sinais de contaminação, como turbidez, é necessário identificar todos os microrganismos isolados nos monitoramentos ambientais, teste de esterilidade e nas unidades envasadas e deve se especificar o próprio gênero dos microrganismos para contribuir na determinação das possíveis fontes de contaminação (IN35/2019).
Efeito adverso é definido como qualquer das etapas de fabricação de medicamentos é composta por muitas etapas e é preciso normatizar para que não haja erros durante os processos, são os parâmetros de Boas Práticas de Fabricação. A RDC 658/22 define os requisitos mínimos que devem ser obrigatoriamente considerados no processo da produção, em conformidade com as Boas Práticas de Fabricação de Medicamentos. Essas normas e recomendações, que visam garantir a qualidade do produto farmacêutico produzido e satisfazer as exigências específicas de certos medicamentos estão sujeitas a alterações e adaptações, mas todos os medicamentos devem atender a critérios de eficácia, segurança e qualidade. Dentre os requisitos pré-estabelecidos a serem observados nos locais de produção para reduzir o risco de danos graves para a saúde, decorrentes da contaminação cruzada, as instalações devem ser segregadas com base nas classes dos medicamentos. Alguns produtos altamente ativos, tais como antibióticos, hormônios, e produtos químicos citotóxicos, devem ser fabricados em instalações separadas (LOYD JR et al., 2007; LEONARDI, 2010; BRASIL, 2022).
IMPLANTAÇÃO DO PROCESSO ASSÉPTICO
A implantação do processo asséptico na área de envase de pós injetáveis apresenta uma série de desafios e considerações específicas que devem ser abordados para garantir a integridade e segurança dos produtos farmacêuticos durante todo o processo de fabricação (KRÄMER et al., 2016).
Um dos aspectos cruciais a serem considerados é o design e a adequação das instalações. A área de envase deve ser cuidadosamente projetada para controlar o ambiente e minimizar a possibilidade de contaminação microbiológica. Isso envolve o estabelecimento de sistemas de ventilação adequados, a segregação de áreas e o estabelecimento de fluxos de pessoal bem definidos. Além disso, a sala limpa é um elemento crítico, exigindo um design apropriado e uma manutenção rigorosa para garantir a integridade do processo asséptico (DIXON, 2016).
A seleção e qualificação dos equipamentos também desempenham um papel fundamental na implantação do processo asséptico. É essencial escolher materiais de construção adequados que sejam compatíveis com os produtos farmacêuticos e não apresentem risco de contaminação. Além disso, a validação da limpeza dos equipamentos, a esterilização adequada e a integridade dos sistemas de filtragem são aspectos críticos a serem considerados. A manutenção regular e a capacitação técnica adequada para a operação dos equipamentos também são fundamentais para garantir a sua eficácia ao longo do tempo (KRÄMER; FEDERICI, 2020).
Os equipamentos utilizados no processo asséptico devem ser construídos com materiais que sejam resistentes à corrosão, não reativos com os produtos farmacêuticos e facilmente limpos e desinfetados. Materiais como aço inoxidável de alta qualidade e polímeros de grau farmacêutico são comumente empregados devido às suas propriedades adequadas (AGALLOCO; AKERS, 2019).
Além da seleção adequada de materiais, a validação da limpeza dos equipamentos é uma etapa crucial. É necessário desenvolver e implementar procedimentos robustos de limpeza e desinfecção para garantir que os equipamentos estejam livres de resíduos, incluindo substâncias potencialmente contaminantes, como microrganismos, partículas e produtos químicos. A validação da limpeza envolve a realização de estudos específicos para demonstrar a eficácia do processo de limpeza (KRÄMER et al., 2016).
Outro aspecto crítico é a esterilização adequada dos equipamentos. Dependendo do tipo de equipamento e da sua complexidade, diferentes métodos de esterilização podem ser empregados, como esterilização por calor úmido (autoclavagem), esterilização por calor seco, radiação gama ou filtração esterilizante. A escolha do método de esterilização deve levar em consideração a natureza dos equipamentos e a viabilidade de cada método para garantir a esterilidade do produto final (AGALLOCO; AKERS, 2019).
O treinamento de pessoal é outra consideração importante na implantação do processo asséptico. Os profissionais envolvidos no processo de envase devem ser adequadamente treinados para garantir a execução correta das práticas assépticas. Isso inclui o conhecimento sobre microbiologia, boas práticas de fabricação, técnicas de esterilização, vestimentas adequadas e higiene pessoal. A conscientização sobre a importância da adesão estrita aos procedimentos e protocolos estabelecidos é fundamental para minimizar o risco de contaminação (AGALLOCO; AKERS, 2021).
Além desses aspectos, a validação do processo é uma etapa crítica na implantação do processo asséptico. A validação deve garantir que todas as etapas do processo, desde a preparação até o envase final, sejam realizadas de forma asséptica e segura. Isso envolve a execução de estudos de validação, como o Media Fill, que simulam o processo de envase em condições reais para garantir a eficácia do processo asséptico (DIXON, 2016).
Em resumo, a implantação do processo asséptico na área de envase de pós injetáveis requer atenção cuidadosa aos desafios e considerações específicas envolvidas. A adequação das instalações, a seleção e qualificação dos equipamentos, o treinamento adequado do pessoal e a validação do processo são elementos fundamentais para garantir a qualidade e a segurança dos produtos farmacêuticos finais. Ao abordar esses aspectos de forma eficaz, as empresas farmacêuticas podem mitigar os riscos associados à contaminação microbiológica e garantir a conformidade com as regulamentações governamentais.
AVALIAÇÃO DA CAPACIDADE DE PROCESSO ASSÉPTICO E IMPORTÂNCIA DO MEDIA FILL
O objetivo do Media Fill é verificar a eficácia do processo asséptico em manter a esterilidade do produto final. Durante o teste, são avaliados diversos aspectos críticos, como a execução das práticas assépticas pelos operadores, a integridade do ambiente controlado, a eficácia dos sistemas de filtragem e a capacidade dos procedimentos de limpeza e desinfecção (SOUZA, 2019).
A realização do Media Fill permite identificar e quantificar possíveis pontos de falha no processo asséptico, auxiliando na identificação de melhorias necessárias e na implementação de ações corretivas. É uma oportunidade de avaliar a eficiência dos treinamentos e procedimentos estabelecidos, bem como a validade das decisões relacionadas ao design das instalações e seleção dos equipamentos (DELJEHIER et al., 2019).
A importância do Media Fill reside na sua capacidade de fornecer informações valiosas sobre a integridade do processo asséptico, ajudando a garantir a qualidade e a segurança dos produtos farmacêuticos. Ao simular condições reais de produção, esse teste possibilita a detecção de potenciais riscos de contaminação e a implementação de medidas preventivas antes da produção em escala real (LINDBOE, 2021).
Um dos principais objetivos do Media Fill é detectar pontos de falha no processo asséptico antes que a produção seja realizada em escala real. Ao preencher o equipamento e seguir todas as etapas do processo, incluindo a manipulação dos componentes e o envase propriamente dito, é possível identificar falhas nas práticas assépticas, como a contaminação proveniente de operadores, falhas nos procedimentos de limpeza e desinfecção, deficiências no design das instalações e até mesmo falhas nos sistemas de filtragem (LINDBOE, 2021).
Ao observar e analisar os resultados do Media Fill, é possível identificar possíveis áreas de melhoria e implementar medidas preventivas antes do início da produção em grande escala. Por exemplo, se forem observadas contaminações durante o teste, podem ser realizadas investigações adicionais para determinar a causa e implementar ações corretivas necessárias. Isso permite evitar a ocorrência de contaminação microbiológica em lotes reais de produtos farmacêuticos, prevenindo problemas de qualidade, recalls e impactos negativos à saúde dos consumidores (HALLS, 2016).
O Media Fill também ajuda a garantir a conformidade regulatória. Muitas agências reguladoras, como a Food and Drug Administration (FDA) nos Estados Unidos e a Agência Nacional de Vigilância Sanitária (ANVISA) no Brasil, exigem a realização desse teste como parte dos processos de validação e monitoramento de processos assépticos. A conformidade com essas regulamentações é essencial para a indústria farmacêutica, pois demonstra o compromisso com a segurança e a qualidade dos produtos fabricados (SOUZA, 2019).
Além disso, o Media Fill é uma exigência regulatória em muitos países e está incorporado em diretrizes e normas, como as Boas Práticas de Fabricação (BPF) e a Farmacopeia. Sua realização regular é uma forma de demonstrar o compromisso da indústria farmacêutica com a garantia da qualidade e a conformidade regulatória (KRÄMER; FEDERICI, 2020).
É importante ressaltar que o Media Fill não é apenas um teste isolado, mas parte de um processo contínuo de avaliação e monitoramento da capacidade do processo asséptico. Os resultados obtidos são analisados e utilizados para aprimorar e aperfeiçoar as práticas assépticas, buscando constantemente a melhoria contínua e a prevenção de riscos associados à contaminação microbiológica.
IDENTIFICAÇÃO DE RISCOS
A identificação de riscos no contexto da implantação do processo asséptico na área de envase de pós injetáveis envolve várias etapas importantes. Primeiramente, é necessário formar uma equipe multidisciplinar composta por profissionais especializados, que possam contribuir com diferentes perspectivas e conhecimentos relevantes para a análise de riscos (YAMAN et al., 2018).
A equipe realiza sessões de brainstorming para identificar eventos ou situações que possam representar riscos ao processo asséptico. Durante essa etapa, é encorajado que todos os membros da equipe expressem suas ideias e considerem diversos cenários possíveis, abrangendo desde falhas no processo asséptico até problemas relacionados a instalações, equipamentos, treinamento e outros aspectos relevantes (AKERS, 2021).
Os riscos identificados são então categorizados e organizados em áreas temáticas, facilitando a visualização e a análise das diferentes fontes potenciais de risco. Nesse sentido, a análise SWOT (forças, fraquezas, oportunidades e ameaças) é uma ferramenta valiosa, permitindo avaliar tanto os fatores internos, como as forças e fraquezas do processo asséptico, quanto os fatores externos, como oportunidades e ameaças que possam influenciar o projeto (GILLINGS et al., 2021).
Uma técnica comumente utilizada para identificar riscos é a análise de causa e efeito, também conhecida como diagrama de Ishikawa ou diagrama de espinha de peixe. Essa abordagem consiste em identificar as principais categorias de causas que podem levar a cada risco identificado e desdobrá-las em subcategorias. Essa análise ajuda a compreender as relações de causa e efeito, bem como a identificar medidas preventivas ou corretivas que possam ser implementadas (GILLINGS et al., 2021).
Durante a identificação de riscos, também é importante revisar a documentação existente, como relatórios de inspeção, registros de incidentes e procedimentos operacionais padrão (POP’s). Esses documentos podem fornecer informações valiosas sobre riscos anteriores ou situações problemáticas, auxiliando na identificação de riscos potenciais (CARREZ et al., 2018).
Após identificar os riscos, é fundamental avaliar a probabilidade de ocorrência e o impacto potencial de cada um deles. Essa avaliação pode ser feita de forma qualitativa, atribuindo classificações como baixo, médio ou alto, ou de forma quantitativa, utilizando escalas numéricas. Essa análise ajuda a priorizar os riscos mais significativos e a definir estratégias de mitigação apropriadas. Algumas técnicas e suas principais podem ser vistas na Tabela 1 construída a partir dos trabalhos de Carrez et al., (2018) e Deljehier et al., (2019).
Quadro 1 – Métodos que podem utilizados na identificação de riscos em processos assépticos.
| Técnica | Características |
| Brainstorming | Reunião de equipe para gerar ideias livremente e em grupo. |
| Encoraja a participação de todos os membros da equipe. | |
| Estimula a criatividade e o pensamento inovador. | |
| Análise SWOT | Avalia as forças, fraquezas, oportunidades e ameaças do processo asséptico. |
| Identifica fatores internos e externos que podem afetar o projeto. | |
| Permite uma visão abrangente do ambiente interno e externo. | |
| Análise de causa e efeito (FMEA) | Também conhecida como diagrama de Ishikawa ou diagrama de espinha de peixe. |
| Identifica as principais categorias de causas que levam aos riscos identificados. | |
| Permite analisar as relações de causa e efeito entre as causas e os riscos. | |
| Ajuda na identificação de medidas preventivas e corretivas. | |
| Revisão de documentação | Análise de relatórios de inspeção, registros de incidentes e procedimentos operacionais padrão (POPs). |
| Identifica riscos anteriores ou situações problemáticas registradas. | |
| Fornecer informações valiosas para identificar riscos potenciais. | |
| Avaliação de probabilidade | Avalia a probabilidade de ocorrência de cada risco identificado. |
| Pode ser feita qualitativamente ou quantitativamente. | |
| Ajuda a priorizar os riscos com base na sua probabilidade. | |
| Avaliação de impacto | Avalia o impacto potencial de cada risco identificado. |
| Pode ser feita qualitativamente ou quantitativamente. | |
| Ajuda a priorizar os riscos com base no seu impacto. |
Fonte: Carrez et al., (2018) e Deljehier et al., (2019).
É importante ressaltar que a identificação de riscos é um processo contínuo e dinâmico. À medida que o projeto avança e novas informações se tornam disponíveis, é necessário revisar e atualizar a lista de riscos identificados. A colaboração e a comunicação constantes entre a equipe são essenciais para garantir uma identificação abrangente e precisa dos riscos relacionados à implantação do processo asséptico.
AVALIAÇÃO DOS RISCOS IDENTIFICADOS
A avaliação dos riscos identificados no contexto do Media Fill é essencial para a gestão eficaz do projeto de implantação do processo asséptico na área de envase de pós injetáveis. Essa avaliação envolve a consideração da probabilidade de ocorrência de cada risco e o impacto potencial que ele pode ter no processo asséptico e na qualidade do produto final (DELJEHIER et al., 2019).
A probabilidade de ocorrência de um risco está relacionada à sua chance de se materializar durante a execução do processo asséptico. Essa probabilidade pode ser avaliada qualitativamente ou quantitativamente, dependendo da disponibilidade de dados e informações. Por exemplo, em uma avaliação qualitativa, os riscos podem ser classificados como baixa, média ou alta probabilidade de ocorrência. Já em uma avaliação quantitativa, podem ser utilizadas escalas numéricas para atribuir valores de probabilidade (LINDBOE, 2021).
O impacto potencial de um risco refere-se às consequências negativas que sua ocorrência pode ter no projeto, no processo asséptico ou na qualidade dos produtos. O impacto pode variar de leve a severo e deve ser avaliado considerando aspectos como segurança do paciente, conformidade regulatória, reputação da empresa, custos financeiros e impactos operacionais. Uma avaliação qualitativa ou quantitativa do impacto pode ser realizada para atribuir uma classificação ou pontuação correspondente (AKERS et al., 2016).
A classificação e priorização dos riscos são etapas cruciais na gestão eficaz do projeto. A classificação permite agrupar os riscos em categorias ou níveis de gravidade, facilitando a compreensão e a comunicação dos riscos para a equipe e as partes interessadas envolvidas no projeto. Isso ajuda a direcionar os esforços de mitigação e recursos para os riscos mais relevantes e críticos (KRÄMER; FEDERICI, 2020).
A classificação dos riscos envolve agrupá-los em categorias ou níveis de gravidade, de acordo com sua importância e impacto potencial. Essa classificação facilita a compreensão e a comunicação dos riscos para a equipe e as partes interessadas envolvidas no projeto. Ao agrupar os riscos em categorias, é possível identificar padrões e tendências, o que auxilia na análise global dos riscos e na tomada de decisões estratégicas (DIXON, 2016).
A categorização dos riscos também ajuda a estabelecer uma linguagem comum entre os membros da equipe, o que é fundamental para uma comunicação clara e eficaz. Ao utilizar categorias ou níveis de gravidade predefinidos, todos têm uma compreensão compartilhada dos riscos e podem concentrar seus esforços em áreas específicas (AGALLOCO; AKERS, 2021).
Além disso, a classificação dos riscos permite identificar os riscos mais relevantes e críticos para o projeto. Ao atribuir uma classificação ou pontuação a cada risco, é possível determinar quais têm maior probabilidade de ocorrer e impacto mais significativo. Essa priorização é importante porque nem todos os riscos têm a mesma importância e requerem a mesma atenção (CARREZ et al., 2018).
A priorização dos riscos, por sua vez, permite estabelecer uma ordem de atuação com base na importância e urgência de cada risco. Riscos com maior probabilidade de ocorrência e impacto mais significativo devem receber maior atenção e ser tratados prioritariamente. Dessa forma, a gestão eficaz dos riscos concentra-se nos aspectos mais críticos e reduz o potencial de ocorrência de eventos indesejados que possam afetar o sucesso do projeto e a segurança dos produtos (YAMAN, 2018).
Por fim, de forma resumida, ao classificar e priorizar os riscos, a equipe pode desenvolver estratégias de mitigação adequadas. Isso pode incluir a definição de ações corretivas específicas, a implementação de controles de qualidade adicionais, a alocação de recursos adequados e o monitoramento contínuo para garantir a eficácia das medidas de mitigação implementadas.
MITIGAÇÃO DOS RISCOS
A mitigação de riscos no contexto do Media Fill envolve a implementação de estratégias e medidas para reduzir a probabilidade de ocorrência e o impacto potencial dos riscos identificados. Essas medidas visam prevenir problemas e garantir a segurança e a qualidade dos processos assépticos e dos produtos farmacêuticos. Aqui estão algumas estratégias e medidas comuns utilizadas para mitigar os riscos no Media Fill (YAMAN, 2018; KRÄMER; FEDERICI, 2020):
- Controles de qualidade: A implementação de controles de qualidade adicionais é uma medida importante para mitigar riscos. Isso pode incluir a realização de testes microbiológicos e físico-químicos rigorosos nos produtos e no ambiente asséptico, antes, durante e após o processo de envase. Os controles de qualidade garantem a detecção precoce de possíveis problemas e permitem a adoção de medidas corretivas imediatas.
- Modificações no processo: Com base nos riscos identificados, podem ser necessárias modificações no processo asséptico para reduzir os riscos. Isso pode envolver a adoção de novas tecnologias ou equipamentos que ofereçam maior segurança e confiabilidade. Por exemplo, pode-se optar por sistemas de fechamento de embalagens mais avançados ou introduzir processos automatizados para minimizar a exposição do produto ao ambiente externo.
- Treinamento adicional do pessoal: A capacitação adequada dos operadores é fundamental para mitigar os riscos no Media Fill. É importante fornecer treinamento contínuo sobre práticas assépticas corretas, procedimentos operacionais padrão (POPs), uso de equipamentos e técnicas de limpeza e desinfecção. O treinamento adicional do pessoal ajuda a minimizar erros humanos, garantindo a execução adequada do processo asséptico.
- Monitoramento contínuo: O monitoramento contínuo é uma medida preventiva importante para identificar e corrigir problemas rapidamente. Isso pode envolver a implementação de sistemas de monitoramento de pressão diferencial, temperatura, umidade e outros parâmetros críticos do ambiente asséptico. Além disso, é essencial realizar auditorias regulares para verificar a conformidade com os procedimentos e as boas práticas de fabricação.
- Análise de causa raiz: Em caso de ocorrência de riscos identificados, é importante realizar uma análise de causa raiz para entender as causas subjacentes e implementar ações corretivas eficazes. A análise de causa raiz busca identificar os fatores que contribuíram para o evento indesejado e desenvolver medidas preventivas para evitar recorrências.
- Revisões periódicas: A revisão periódica dos riscos identificados e das medidas de mitigação implementadas é fundamental para manter um processo asséptico robusto e atualizado. À medida que o projeto avança e novas informações se tornam disponíveis, é importante avaliar regularmente os riscos e as medidas de mitigação para garantir sua eficácia contínua.
Halls (2016) aponta que as revisões periódicas são fundamentais para manter um processo asséptico robusto e atualizado. À medida que o projeto avança e novas informações se tornam disponíveis, é necessário avaliar regularmente os riscos identificados e as medidas de mitigação implementadas. Isso garante que as estratégias sejam eficazes e atualizadas para enfrentar os desafios em constante evolução.
A mitigação de riscos no contexto do Media Fill envolve a implementação de várias estratégias e medidas, como controles de qualidade adicionais, modificações no processo, treinamento adicional do pessoal, monitoramento contínuo, análise de causa raiz e revisões periódicas. Essas ações preventivas visam reduzir a probabilidade de ocorrência e o impacto potencial dos riscos identificados, garantindo a segurança e a qualidade dos processos assépticos e dos produtos farmacêuticos (AGALLOCO; AKERS, 2019).
BENEFÍCIOS DA ANÁLISE DE RISCO NA GESTÃO DE PROJETOS
A incorporação da análise de risco na gestão do projeto de implantação do processo asséptico no contexto do Media Fill traz diversos benefícios significativos. Primeiramente, a análise de risco permite a redução de falhas no processo. Ao identificar antecipadamente os possíveis eventos ou situações que podem levar a falhas no processo asséptico, é possível implementar medidas preventivas e corretivas adequadas. Isso resulta em uma maior confiabilidade do processo, reduzindo problemas operacionais, interrupções na produção e retrabalhos, o que, por sua vez, aumenta a eficiência e a produtividade do projeto (KRÄMER; FEDERICI, 2020).
Além disso, a análise de risco desempenha um papel fundamental na garantia da qualidade do produto final. Ao identificar e avaliar os riscos potenciais de contaminação, é possível implementar controles e procedimentos adequados para evitar a introdução de impurezas ou micro-organismos nocivos. Isso resulta em produtos finais de maior qualidade, reduzindo o risco de rejeição e garantindo a segurança e a eficácia dos medicamentos produzidos (KRÄMER et al., 2016).
Outro benefício importante é a contribuição para a conformidade regulatória. A análise de risco ajuda a identificar e avaliar os riscos relacionados a aspectos regulatórios. Com base nesses riscos identificados, é possível implementar medidas que garantam a conformidade com as normas e regulamentações vigentes, como as Boas Práticas de Fabricação (BPF). Isso reduz o risco de não conformidade, auditórias negativas e penalidades regulatórias, contribuindo para a reputação da empresa e sua posição no mercado (SOUZA, 2019).
Souza (2019) aponta ainda que a análise de risco também desempenha um papel crucial na tomada de decisão embasada durante o projeto de implantação do processo asséptico. Ao ter uma compreensão clara dos riscos envolvidos, a equipe de projeto pode tomar decisões mais informadas e estratégicas. Isso inclui direcionar recursos para áreas de maior risco e prioridade, aumentando a eficácia da gestão do projeto, reduzindo a incerteza e maximizando as chances de sucesso.
Por fim, a análise de risco promove a melhoria contínua do processo asséptico. Não se trata apenas de uma etapa inicial do projeto, mas sim de um processo contínuo ao longo do ciclo de vida do processo. A equipe pode revisar e atualizar regularmente a análise de risco à medida que novas informações se tornam disponíveis, novos riscos são identificados ou as circunstâncias mudam. Isso permite uma melhoria contínua do processo, garantindo que os riscos sejam monitorados e gerenciados adequadamente ao longo do tempo. (LINDBOE, 2021)
Em resumo, a incorporação da análise de risco na gestão do projeto de implantação do processo asséptico no contexto do Media Fill traz benefícios que vão desde a redução de falhas até a garantia da qualidade do produto final, a conformidade regulatória e a tomada de decisão embasada. Essa abordagem promove a segurança, a eficácia e o sucesso do processo asséptico, proporcionando resultados de alta qualidade na indústria farmacêutica.
MATERIAIS E MÉTODOS
Trata-se de um estudo de análise de risco utilizando a ferramenta de gestão FMEA (Failure Mode and Effect Analysis), para gerenciar os principais riscos que são observados na execução do projeto de implantação do processo asséptico na área de envase. Foram utilizados dados hipotéticos para a análise de risco.
Foram apresentados os dados principais na implantação do processo asséptico na tabela FMEA e realizada as avaliações necessárias para o estudo.
A utilização da FMEA oferece uma abordagem estruturada para avaliação do projeto e corrobora com a eficiente prevenção de problemas na implantação do projeto. E esta ferramenta foi selecionada para este estudo por permitir a identificação e análise das possibilidades de falhas além de identificar as causas e efeitos sobre o sistema.
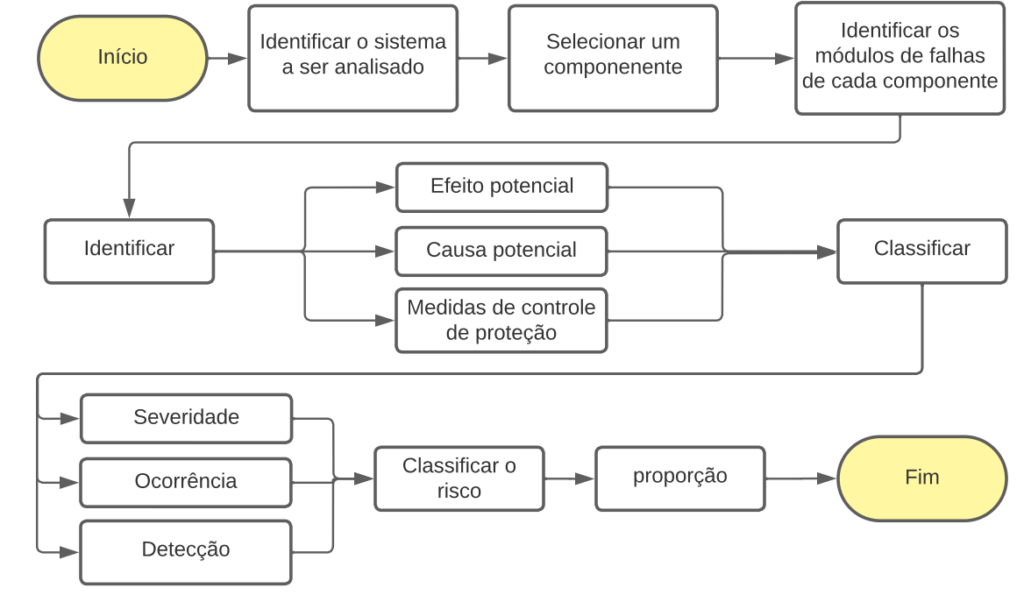
O preenchimento segue a ordem do fluxograma das atividades conforme a Figura 2. A sequência metodológica apresentada para um estudo de análise de risco no processo de Media Fill começou pela identificação do sistema a ser analisado. Este foi um passo fundamental que estabeleceu o escopo da análise de risco, definindo o processo ou procedimento que foi estudado, neste caso, o processo Media Fill.
Em seguida, um componente específico foi selecionado para análise. Em um processo complexo, há várias partes ou componentes que podem ser analisados. Por exemplo, no processo Media Fill, os componentes poderiam ser as diferentes etapas do processo, como a preparação do meio, a inoculação, a incubação, a inspeção, entre outros.
Posteriormente, os modos de falha de cada componente foram identificados. Modos de falha referem-se às maneiras específicas pelas quais um componente pode falhar ou não funcionar como pretendido. Para cada modo de falha, uma análise individual foi realizada.
Em seguida, para cada modo de falha, o efeito potencial, a causa potencial e as medidas de controle de proteção foram identificadas. O efeito potencial refere-se ao impacto que a falha teria no processo global. A causa potencial refere-se ao que poderia potencialmente levar a essa falha. As medidas de controle de proteção são os procedimentos ou políticas em vigor para prevenir a ocorrência dessa falha.
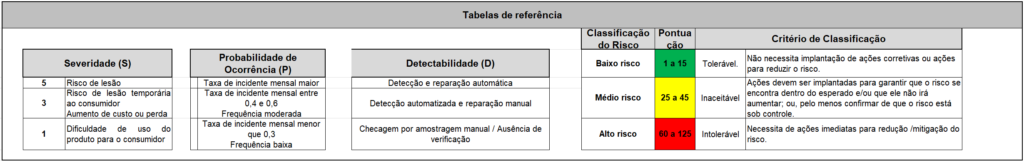
Posteriormente, a severidade, ocorrência e detecção de cada falha foram classificadas. A severidade refere-se à gravidade do impacto caso a falha ocorra. A ocorrência é uma estimativa da frequência com que essa falha pode acontecer. A detecção é uma avaliação de quão provável é que a falha seja descoberta antes que cause um problema significativo.
Com base nessas classificações, o risco associado a cada modo de falha foi então classificado. Essa classificação de risco proporcionou uma compreensão do potencial de dano que cada falha poderia ter e quão crítica era a necessidade de medidas de controle.
Finalmente, proporções adequadas para minimizar ou eliminar os riscos foram propostas. Essas proporções puderam incluir medidas de controle adicionais, treinamento, mudanças no processo, entre outros.
Figura 02. Fluxograma das atividades adotadas para a condução da avaliação usando FMEA (Failure Mode and Effect Analysis).
RESULTADOS E DISCUSSÃO
A Tabela 2 apresenta a planilha de avaliação de risco na simulação de implementação do processo asséptico na área de envase de pós injetáveis.
Tabela 2 – Planilha de avaliação de risco na implementação do processo asséptico na área de envase de pós injetáveis.
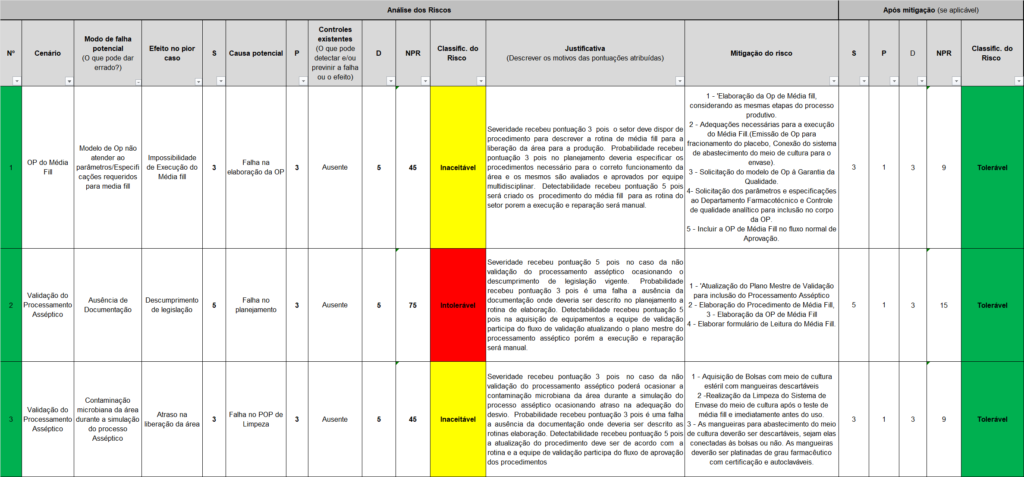
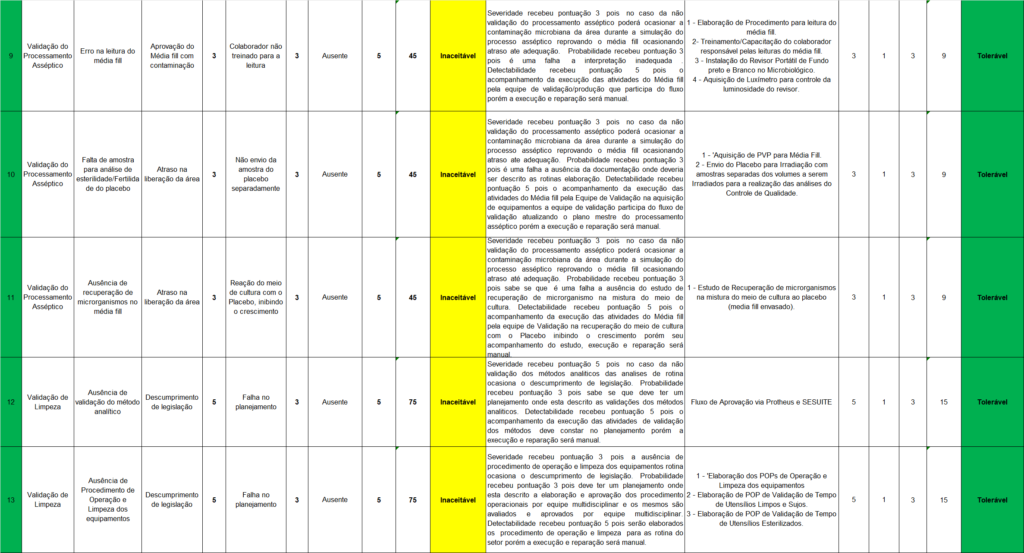
A Tabela 2 traz um exemplo de um estudo de gerenciamento de risco que desenvolveu uma Análise de Modo e Efeitos de Falha (FMEA) (Failure Mode and Effect Analysis), uma metodologia utilizada para identificar e avaliar riscos associados a um processo ou produto, especificamente, o processo de envase de produtos injetáveis neste caso.
Na tabela, “N°” corresponde a um número sequencial para cada cenário de risco sendo analisado. “Cenário” detalha o cenário específico em avaliação. Dentro do contexto de envase de produtos injetáveis, pode ser uma fase específica do processo de produção, tal como o “Enchimento do frasco”.
“Modo de falha potencial” descreve o que poderia sair errado em cada cenário, tal como uma “Contaminação do produto durante o processo de enchimento”. A coluna “S” representa a severidade da falha, que é geralmente avaliada numa escala de 1 (baixa severidade) a 10 (alta severidade).
“Efeito no pior caso” descreve o cenário mais grave caso ocorra a falha, tal como “Retirada do produto do mercado e danos à saúde do paciente”. “Causa potencial” refere-se às possíveis causas do modo de falha, como uma “Falha nos procedimentos de higiene”.
A coluna “P” estima a probabilidade de ocorrência da falha, também medido numa escala de 1 (baixa probabilidade) a 10 (alta probabilidade). “Controles existentes” descreve as medidas atuais para prevenir a falha, como “Procedimentos de limpeza e desinfecção”.
“D” representa a detectabilidade da falha, medida em uma escala de 1 (fácil de detectar) a 10 (difícil de detectar). “NPR” refere-se ao Número Prioritário de Risco, calculado pela multiplicação da severidade, probabilidade e detectabilidade. Este número é utilizado para classificar os riscos e priorizar ações corretivas.
“Classificação do risco” é uma classificação do risco baseada no NPR, podendo ser categorizada como “baixo”, “médio” ou “alto”. “Justificativa” provê uma explicação para a classificação do risco e a necessidade de ação corretiva.
A, “Mitigação do risco” descreve as ações planejadas para minimizar o risco. A efetividade da FMEA (Failure Mode and Effect Analysis) reside na garantia de que cada fase do processo seja examinada de forma completa e que todas as ações corretivas planejadas sejam implementadas e monitoradas quanto à sua eficácia (MCIVER, 2019). Além disso, a análise FMEA deve ser um processo contínuo, revisto regularmente à medida que os processos mudam ou novos riscos são identificados.
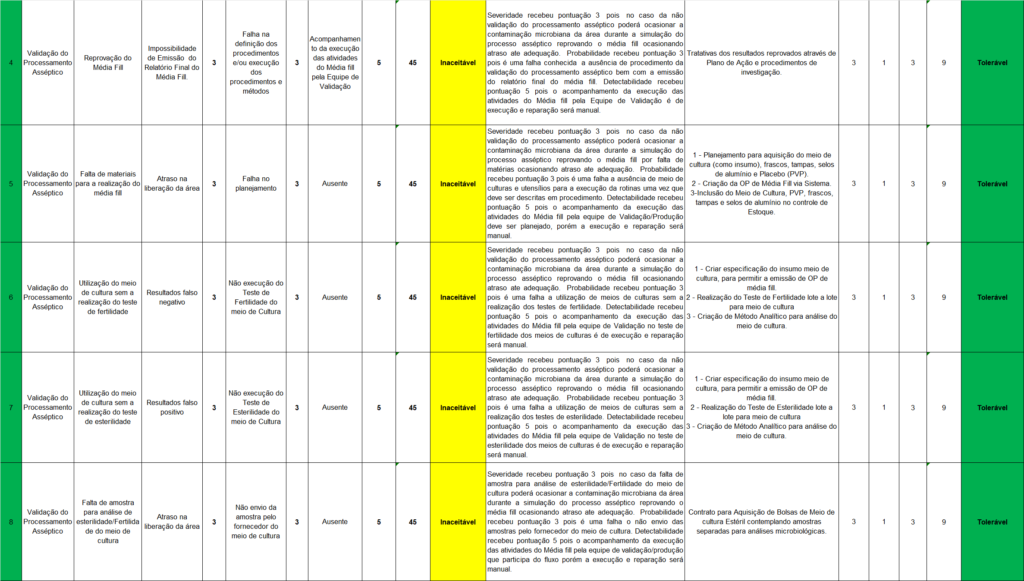
Após detalhar as variáveis que compõem a Tabela 2, que apresenta 18 cenários de riscos, observa-se que apenas um dos processos descritos tem uma classificação de risco definida como Intolerável. Este cenário corresponde à falta de documentação na validação do processo asséptico, uma infração regulamentar classificada com uma severidade de 5. Para mitigar esse risco, sugere-se a atualização do plano de validação para incluir o processo asséptico, a elaboração do procedimento Media Fill, além da criação de um formulário de leitura.
A Tabela 2 indica que 17 cenários foram identificados como tendo um nível de risco “Inaceitável”, englobando aspectos como validação de limpeza, processamento asséptico e o modelo de protocolo de validação de processo. Estes cenários, todos com probabilidade de ocorrência de nível 3, sugerem que existem falhas sistemáticas que precisam ser abordadas para garantir a qualidade e a segurança do processo de envase.
Uma importante observação é que, após a implementação de medidas de mitigação, houve uma redução significativa na probabilidade de ocorrência e na detectabilidade de alguns riscos. Notavelmente, o texto aponta que a severidade de um problema se mantém constante, indicando que a ocorrência de tal problema seria grave, independentemente das causas.
Por meio da Tabela 2, também se nota que a maioria das causas potenciais para cada cenário está relacionada à falha de planejamento, o que implica problemas na gestão da área. Assim, sugere-se a adoção de medidas para melhorar o planejamento e a gestão das tarefas. A menção a atrasos na liberação da área e soluções propostas, como a aquisição de PVP para Media Fill e o envio do placebo para irradiação com amostras separadas, aponta para a necessidade de revisão e aprimoramento dos procedimentos existentes.
Em termos gerais, a análise contida na Tabela 2 fornece uma orientação valiosa para a área de envase de pós-injetáveis, permitindo a identificação de problemas potenciais e a proposição de métodos para reduzir os riscos associados. O aspecto da documentação, bem como as questões operacionais, são considerados.
Os dados da Tabela 2 mostram a necessidade de implementar as ações de mitigação identificadas. Para que isso ocorra de maneira efetiva, é importante considerar a viabilidade econômica e técnica de cada ação. Essa abordagem garante que os esforços de melhoria não sejam apenas teóricos, mas também práticos e realizáveis, tendo em vista a realidade da empresa e os recursos disponíveis.
É crucial destacar que a avaliação de risco deve ser considerada uma fase inicial no processo de melhoria contínua de qualquer organização.
Esta análise, como evidenciado pelo texto, revela que uma grande proporção dos riscos identificados está vinculada à operação dos profissionais. Esta constatação é significativa, pois indica que o foco para a mitigação desses riscos não necessariamente exige um grande investimento em tecnologia ou equipamentos novos, mas ressalta a necessidade de capacitação dos funcionários.
Isto sugere que a implementação de treinamentos consistentes e abrangentes pode ser uma estratégia eficaz para reduzir os riscos inerentes à operação. Assegurar que os profissionais estejam bem informados sobre os processos, normas para melhorar a qualidade e a segurança do trabalho, ao mesmo tempo que minimiza a probabilidade de erros operacionais.
Adicionalmente, a padronização das operações é outro ponto crucial que o texto enfatiza. Processos padronizados não apenas reduzem a variabilidade e a chance de erros, mas também facilitam o treinamento dos funcionários, pois estabelecem um conjunto claro e uniforme de procedimentos a serem seguidos. A padronização contribui ainda para a eficiência geral, uma vez que os processos são mais facilmente replicáveis e previsíveis (DIXON, 2016).
Por fim, é importante ressaltar que, mesmo com treinamento e padronização, a avaliação de risco deve ser uma atividade recorrente. Isso porque os processos e as tecnologias evoluem, e novos riscos podem surgir. Portanto, uma avaliação de risco periódica permite à organização manter-se atualizada e proativa na identificação e mitigação de potenciais riscos.
Conclusão
A implantação do processo asséptico na área de envase de pós injetáveis, auxiliada pela análise de risco, é de suma importância para a segurança e a qualidade dos produtos farmacêuticos. Com a adoção de estratégias e medidas de mitigação de riscos, incluindo controles de qualidade adicionais, modificações nos procedimentos, treinamento do pessoal, monitoramento contínuo e análises de causa-raiz, é possível minimizar a probabilidade e o impacto potencial de eventos adversos. Essas medidas não apenas diminuem a incidência de falhas, mas também garantem a qualidade do produto final e a conformidade com as regulamentações, trazendo benefícios significativos para a indústria farmacêutica.
Além disso, a inclusão da análise de risco no gerenciamento do projeto para a implementação do processo asséptico no âmbito do Media Fill proporciona uma abordagem proativa na identificação, avaliação e mitigação dos riscos. Empregando técnicas como brainstorming, análise SWOT e análise de causa e efeito, os riscos potenciais podem ser identificados, sua probabilidade e impacto podem ser avaliados e estratégias de mitigação apropriadas podem ser desenvolvidas. Isso permite uma tomada de decisão mais informada e fundamentada, canalizando recursos e esforços para as áreas de maior risco e prioridade.
A mitigação de riscos no contexto do Media Fill leva a uma redução de falhas no processo asséptico, resultando em maior confiabilidade e eficiência operacional. A qualidade do produto final é assegurada através da implementação de controles de qualidade rigorosos e ajustes nos procedimentos para minimizar os riscos de contaminação. Essas medidas têm como objetivo garantir que os produtos farmacêuticos sejam seguros, eficazes e estejam em conformidade com as normas e regulamentações aplicáveis (LINDBOE, 2021).
A conformidade regulatória é um aspecto crucial para a indústria farmacêutica. A análise de risco desempenha um papel vital na identificação e avaliação dos riscos associados aos requisitos regulatórios, possibilitando a implementação de medidas para garantir a conformidade com as Boas Práticas de Fabricação (BPF) e outras regulamentações relevantes. Isso ajuda a evitar não conformidades, penalidades regulatórias e danos à reputação da empresa, fortalecendo sua posição no mercado.
A análise de risco no contexto do Media Fill fomenta a melhoria contínua do processo asséptico. Por meio de revisões regulares e atualizações na análise de risco, novos riscos são identificados e novas informações são incorporadas, permitindo o aprimoramento contínuo das estratégias de mitigação. Isso garante que os riscos sejam efetivamente monitorados e que medidas preventivas e corretivas sejam implementadas quando necessário, assegurando a segurança e a qualidade dos produtos farmacêuticos em todas as etapas do processo.
Em resumo, a análise de risco desempenha um papel fundamental na gestão do projeto de implementação do processo asséptico no contexto do Media Fill. Ao identificar, avaliar e mitigar riscos, é possível reduzir falhas, garantir a qualidade do produto final e alcançar a conformidade regulatória. A abordagem proativa da análise de risco traz benefícios significativos para a indústria farmacêutica, promovendo segurança, eficácia e excelência no processo asséptico de envase de pós injetáveis.
REFERÊNCIAS:
AGALLOCO, J.; AKERS, J. Risk assessment and mitigation in aseptic processing. In: Parenteral Medications. CRC Press, 2019. p. 829-839.
AGALLOCO, J.; AKERS, J. Aseptic Processing of Sterile Dosage Forms. In: Handbook of Validation in Pharmaceutical Processes. 2021. p. 443-456.
AKERS, J. et al. Highly automated isolator-based vaccine filling—a case study. In: Advanced Aseptic Processing Technology. CRC Press, 2016. p. 407-415.
AKERS, M. J. Sterile drug products: formulation, packaging, manufacturing, and quality. CRC Press, 2016.
AKERS, M. J. Good Aseptic Practices: Education and Training of Personnel Involved in Aseptic Processing. In: Aseptic Pharmaceutical Manufacturing II. CRC Press, 2020. p. 181-221.
ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS. ABNT NBR IEC 31010: Gestão de riscos – Técnicas para o processo de avaliação de riscos – Diretrizes. Rio de Janeiro, 2012.
ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS. ABNT NBR IEC 31000: Gestão de riscos – Diretrizes. Rio de Janeiro, 2018.
BLACK, K. Causes of project failure: a survey of professional engineers. PM Network, p. 21-24, 1996.
BRASIL. Resolução RDC nº 658, de 30 de março de 2022. Diário Oficial da União, Brasília, DF, 31 mar. 2022. Disponível em: https://www.in.gov.br/en/web/dou/-/resolucao-rdc-n-658-de-30-de-marco-de-2022-389846242. Acesso em: 23 jun. 2023.
BRASIL. AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA. Consulta Pública nº 39, de 29 de abril de 2010. Diário Oficial da União, Brasília, DF, 30 abr. 2010. Disponível em: <www.anvisa.gov.br>. Acesso em: 23 jun. 2023.
BRASIL. AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA. Guia da Qualidade para Sistemas de Tratamento de Ar e Monitoramento Ambiental na Indústria Farmacêutica. Brasília, DF, 2013.
BRASIL. TRIBUNAL DE CONTAS DA UNIÃO. Secretaria Geral de Controle Externo. Referencial básico de gestão de riscos. Brasília, 2018.
CARREZ, L. et al. Qualification and performance evaluation of an automated system for compounding injectable cytotoxic drugs. Pharmaceutical Technology in Hospital Pharmacy, v. 3, n. 3, p. 165-175, 2018.
CEDGEI – COMISSÃO EUROPEIA DIRECÇÃO GERAL DAS EMPRESAS E DA INDUSTRIA. Eudralex – Gestão de Risco de Qualidade. Vol. 4, 2008.
CICMIL, S. Understanding project management practice through interpretative and critical research perspectives. Project Management Journal, v. 37, n. 2, 2006.
CLELAND, D. I. Project management: strategic design and implementation. 2nd ed., 1994.
COX, L. et al. Allergen immunotherapy practice in the United States: guidelines, measures, and outcomes. Annals of Allergy, Asthma & Immunology, v. 107, n. 4, p. 289-299, 2016.
DELJEHIER, T. et al. Simulation program of a cytotoxic compounding robot for monoclonal antibodies and anti-infectious sterile drug preparation. Journal of Oncology Pharmacy Practice, v. 25, n. 8, p. 1873-1890, 2019.
DIXON, A. M. Process Simulations (Media Fills). In: Environmental Monitoring for Cleanrooms and Controlled Environments. CRC Press, 2016. p. 115-134.
GILLINGS, N. et al. Guideline on current good radiopharmacy practice (cGRPP) for the small-scale preparation of radiopharmaceuticals. EJNMMI Radiopharmacy and Chemistry, v. 6, n. 1, p. 1-22, 2021.
HALLGREEN, M.; MAANINEN-OLSSON, E. Deviations, ambiguity and uncertainty in a project-intensive organization. Project Management Journal, v. 36, n. 3, p. 17-26, 2005.
HALLS, N. Media Fills and Their Applications. In: Microbiological Contamination Control in Pharmaceutical Clean Rooms. CRC Press, 2016. p. 65-96.
HARTMAN, F. Preparing the mind for dynamic management. International Journal of Project Management, v. 26, p. 258-267, 2008.
HARTMAN, F.; ASHRAFI, R. Project Management in the information technology and information systems industry. Project Management Journal, v. 33, n. 3, p. 5-15, 2002.
IN – INSTRUÇÃO NORMATIVA, nº 35, de 21 de Agosto de 2019. Dispõe sobre as Boas Práticas de Fabricação complementares a Medicamentos Estéreis – Entidade – ANVISA.
INTERNATIONAL CONFERENCE ON HARMONIZATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE. ICH Q9 – Quality Risk Management. 2005.
JAAFARI, A. Project Management in the age of complexity and change. Project Management Journal, v. 34, n. 4, p. 47-57, 2003.
KPMG. KPMG’s International 2002-2003. Programme management survey, 2002.
KRÄMER, I.; FEDERICI, M. Implementation and microbiological stability of dose-banded ganciclovir infusion bags prepared in series by a robotic system. European Journal of Hospital Pharmacy, v. 27, n. 4, p. 209-215, 2020.
KRÄMER, I. et al. Media-fill simulation tests in manual and robotic aseptic preparation of injection solutions in syringes. Journal of Oncology Pharmacy Practice, v. 22, n. 2, p. 195-204, 2016.
KREINER, K. In search of relevance: project management in drifting environments. Scandinavian Journal of Management, v. 11, n. 4, p. 335-346, 1995.
LEONARDI, E. RDC 17/10, sobre boas práticas de fabricação de medicamentos, comentada. 2010. Disponível em: https://ictq.com.br/industria-farmaceutica/926-rdc-17-10-sobre-boas-praticas-defabricacao-de-medicamentos.
LEYBOURNE, S. A. The changing bias of project management research: a consideration of the literatures and an application of extant theory. Project Management Journal, v. 38, n. 1, p. 62-73, 2007.
LINDBOE, W. G. Validation of container preparation processes. In: Handbook of Validation in Pharmaceutical Processes. CRC Press, 2021. p. 517-525.
LOBO, R. N. Gestão da Qualidade. 2nd ed., 2020.
LOYD, J. R. A. et al. Formas farmacêuticas e sistemas de liberação de fármacos. 8th ed., 2007.
MARQUES JUNIOR, L. J. Uma contribuição para melhoria do planejamento de empreendimentos de construção em organizações públicas. Dissertação (Mestrado) – Escola Politécnica da Universidade de São Paulo, 2000.
MAYLOR, H. Beyond the gantt chart: project management moving on. European Management Journal, v. 19, p. 92-100, 2001.
MCIVER, D. Microbiological Aspects of Pharmaceutical Aseptic Processing in the Compounding Pharmacy. In: Good Manufacturing Practices for Pharmaceuticals. CRC Press, 2019. p. 301-310.
MENEZES, L. C. Gestão de Projetos. CATHO, educação executiva, 2009.
MIDLER, C. Project management for intensive, innovation-based strategies: new challenges for the 21st century. In: SLEVIN, D. P.; CLELAND, D. I.; PINTO, J. K. (Eds.). The frontiers of project management research, 2002.
MILLS, B. et al. Managing technological innovation projects: the quest for a universal language. In: SLEVIN, D. P.; CLELAND, D. I.; PINTO, J. K. (Eds.). The Frontiers of Project Management Research, 2002.
MURIITHI, N.; CRAWFORD, L. Approaches to project management in Africa: implications for international development projects. International Journal of Project Management, v. 21, p. 309-319, 2003.
NAKASHIMA, D. T. V.; CARVALHO, M. M. Identificação de riscos de projetos de TI. Sem data.
PACKENDORFF, J. Inquiring into the temporary organization: new directions for Project management research. Scandinavian Journal of Management, v. 11, n. 4, 1995.
PROJECT MANAGEMENT INSTITUTE. A guide to the project management body of knowledge. 2004.
PROJECT MANAGEMENT INSTITUTE. Um guia do conhecimento em gerenciamento de projetos (Guia PMBOK). 6th ed., 2017.
SALLES JR., C. A. C. et al. Gerenciamento de riscos em projetos. 2ª ed., 2010.
SHENHAR, A. J.; DVIR, D. Reinventing project management: the diamond approach to successful growth & innovation. 2007.
STOECKLIN, W. G. Aseptic Technique: A Goal to Strive For. In: Advanced Aseptic Processing Technology. CRC Press, 2016. p. 3-15.
VAN DER WALT, D.; SPIES, E. The real work of project management in the new economy: a case of software development projects. South African Journal of Information Management, v. 4, n. 2, p. 1-14, 2002.
WILLIAMS, T. Assessment of risk management maturity—closing the gap. Project Management Journal, v. 36, n. 4, 2005.
WILLIAMS, T. M. Post-project reviews to gain effective lessons learned. Project Management Institute, 2007.
1 Discente do Curso de Farmácia da Faculdade FIBRA
2 Professora Doutora do Curso de Farmácia, Faculdade FIBRA
3 Orientadora e Professora Doutora do Curso de Farmácia, Faculdade FIBRA
E-mail: bunaol@hotmail.com