REGISTRO DOI: 10.5281/zenodo.8140802
Gabriel Barbosa Huszcz1,2, Gledison Eric Teixeira 1,
Igor Henrique Rodrigues-Oliveira3, Renan Rodrigues Rocha2,3,
Rafaela de Campos Oliveira2, Paulo Sallarola Takao2,
David Aciole Barbosa1, Daniela Leite Jabes1,
Karine Frehner Kavalco3, Rubens Pasa3,
Fabiano Bezerra Menegidio1,2
Abstract: Fungi of the Malassezia genus are the most prevalent eukaryotic organisms in the natural skin mycobiome of animals and humans. The genus is found in various parts of the body, causing common skin diseases such as pityriasis versicolor and seborrheic dermatitis, and its high adaptability to the human body and prevalence among other eukaryotes makes it unique. However, even with eighteen known species and genomes available, studies on their phylogeny and basic genomic information are still scarce. To this day, only nine species have their mitochondrial genome described and assembled. In this study, we describe for the mtDNA of Malassezia vespertilionis, Malassezia dermatis, Malassezia nana, Malassezia equina, Malassezia caprae and six new mitogenomes of Malassezia pachydermatis. In addition, to better understand the phylogenetic relationships, we mined 35 mitogenomes belonging to 9 species of Malassezia, along with the mitogenomes gathered in this study, and reconstructed the phylogeny based on mtDNA. The results of this study provide genomic variation information and enhance the understanding of the Malassezia genus. They could later deliver highly valuable new insight into data for phylogenetic analysis and population genetics.
Keywords: Mitochondrial genome, Malassezia vespertilionis, Malassezia dermatis, Malassezia nana, Malassezia equina, Malassezia caprae, Malassezia pachydermatis, Phylogeny.
Introduction
Yeasts of the Malassezia genus are present in the microbiota of many animals, including humans, living as commensal organisms on the skin and scalp (Findley et al., 2020). However, in some cases, they can become pathogenic under the influence of predisposing factors that involve both the conditions of the skin microenvironment and changes in the host’s immune system (Theelen et al., 2018; Aykut et al., 2019; Limon et al., 2019; Saunte et al., 2020; Spatz and Richard 2020). The genus groups basidiomycetic, lipophilic (M. pachydermatis) and/or lipodependent yeasts that show asexual reproduction by unipolar or sympodial budding (M. sympodialis) from the parent cell.
Another important feature is the possibility of forming pseudohyphae.
Differentiation of species in the traditional way is done through lipid and fatty acid assimilation tests (biochemical methods), also considering the characteristics of micromorphology. Currently, molecular biology methodologies have been considered of great importance for the taxonomic grouping of the Malassezia genus. The support of molecular tools is very important, since the morphological characteristics between species are very subtle, which can generate doubts and errors. Study of the D1/D2 regions of the 28S ribosomal RNA gene (rRNA28S) and the ITS (Internal Transcribed Spacer) regions have helped in the differentiation of Malassezia species (Batra et al., 2005). In 1996, based on studies based on morphology, physiology and molecular biology, there was a change in the classification of the genus Malassezia in seven species. As of 2002, another seven new species of Malassezia were cited in the literature. Currently, 18 species are recognized for the genus, with only 17 having available genomes (M. arunalokei, M. caprae, M. cuniculi, M. dermatis, M. equina, M. furfur, M. globosa, M. japonica, M. nana, M. obtusa, M. pachydermatis, M. restricta, M. slooffiae, Malassezia sp., M. sympodialis, M. vespertilionis and M. yamatoensis) and 9 having mitochondrial genomes (M. furfur, M. globosa, M. japonica, M. obtusa, M. pachydermatis, M. restricta, M. slooffiae, M. sympodialis and M. yamatoensis).
In this study, we describe the mitochondrial genome of five species of Malasseziales: Malassezia vespertilionis, Malassezia dermatis, Malassezia nana, Malassezia equina and Malassezia caprae, also six new mitogenomes of Malassezia pachydermatis. In addition, to better understand the phylogenetic relationships, we mined 35 mitogenomes belonging to 9 species of Malassezia, along with the mitogenomes gathered in this study, and reconstructed the phylogeny based on mtDNA.
Materials & Methods
Mitogenome assembly and annotation
We obtained Whole Genome Sequencing (WGS) raw data of Malassezia vespertilionis (SRR6206152; Lorch et al., 2018), Malassezia dermatis (DRR043255; Sugita et al., 2002), Malassezia nana (DRR043258; Hirai et al., 2004), Malassezia equina (SRR2136627; Wu et al.,2015) and Malassezia caprae (SRR2132347; Wu et al., 2015) from the Sequence Reads Archive (SRA) NCBI.
The reads were submitted to a workflow previously used and validated in fish mitogenome assemblies by Rocha-Reis et al. (2020), Resende et al. (2020) and Pasa et al. (2021), with minor modifications to adapt to the assembly of fungal mitogenomes. We imported the raw data into the Galaxy Europe platform (https://usegalaxy.eu/) and used NOVOplasty v4.3.1 (Dierckxsens et al.,2017) to assemble the mitochondrial genomes. As a seed, we used the complete mitogenome of M. japonica (KY911090.1). We annotated the sequences obtained on Mitos2 (http://mitos2.bioinf.uni-leipzig.de/; Arab et al., 2017) using Gene Code 3 – Yeast and RefSeq 89 Fungi.
To validate our workflow, we also performed the assembly and annotation of six new mitogenomes from Malassezia pachydermatis WGS libraries (SRR12046913, SRR12005311, SRR12005312, SRR12005313, SRR12005314 and SRR2135029). In addition, the 34 mitogenomes of the 9 species of the Malassezia genus available at the NCBI (M. furfur, M. globosa, M. japonica, M. obtusa, M. pachydermatis, M. restricta, M. slooffiae, M. sympodialis and M. yamatoensis) were annotated according to the procedure described above.
The circular map of the new mitogenomes was drawn using the online software GenomeVx (Conant and Wolfe, 2008). We used the Blast Ring Image Generator (BRIG) (Alikhan et al., 2011) to perform a comparative Blast analysis of available representative Malassezia mitogenomes against our mitogenome pools.
Mitochondrial phylogenetic analysis
To perform the phylogenetic analyses, we manually extracted the sequences of the 15 proteincoding genes (PCGs) of the 11 mitogenomes assembled in this work and added to the dataset the PCGs of the 34 mitogenomes of other Malassezia species available at the NCBI, addition to the Rhodotorula mucilaginous (MF694646.1) mitogenome as an outgroup. We aligned the sequences with MAFFT v7 (Katoh & Standley, 2013), available on the Galaxy Europe online platform (Afgan et al., 2018), and concatenated the alignments in SequenceMatrix v1.8 (Vaidya et al., 2011). We constructed the phylogeny by the Maximum Likelihood (ML) method in the IqTree v2.1.2 software (Nguyen et al., 2015) using 1000 ultra-fast bootstrap repeats and the evolutionary model GTR+F+I+G4 estimated by the IqTree ModelFinder (Kalyaanamoorthy et al., 2017).
Data Description
Characterization of mitochondrial genomes of Malassezia species
The mitogenome of the five Malassezia species are circular DNA molecules with a total length ranging from 27,284 bp to 40,970 bp. Mitogenome sizes varied widely among the five newly assembled mitochondrial genomes, with M. vespertilionis having the largest among the new mtDNA and M. caprae being the smallest mitogenome ever described for the genus. Furthermore, M. equina becomes the second smallest mitogenome described for the genus, followed by the already described M. japonica. However, M. dermatis and M. nana presented a mitogenome with a size close to the average of other species of the genus. Likewise, the new M. pachydermatis mitogenomes were between 35,817 bp and 35,829 bp in size, being slightly larger than the M. pachydermatis strain CBS1879 (KY911092.1) mitogenome previously described. A comparison between the size of the new mitogenomes described in this work and those available in the NCBI database can be seen in Supplementary Tables 1.
The GC concentration of the mitogenome differs among species, which may be affected by mutation bias, selection, and biases of reconstitution-related DNA repair (Li et al., 2018). The low G + C content observed is similar to other fungal mitogenomes in Malassezia (Supplementary Table 1). The GC content in the five mitogenomes ranged from 23.4% to 33.7%, among which the GC content in the genome of M. caprae was the highest.
The base composition of the mitochondrial genome of M. caprae was estimated as 33.8% A, 17.2%C, 16.5% G, and 32.5% T, thus presenting a bias towards AT (66.3%) and the GC content was 33.7%. M. equina presented the composition of 33.4% A, 33.7% T, 16.0% C and 16.1% G, with a bias towards AT (67.1%) and GC content of 32.9%. M. nana presented the composition of 33.6% A, 34.5% T, 15.8% C and 16.1% G, with a bias towards AT (68.1%) and GC content of 31.9%.M. vespertilionis presented the composition of 36.8% A, 39.8% T, 11.5% C and 11.8% G, with a bias towards AT (76.6%) and GC content of 23.4%. Finally, M. dermatis presented a composition of 34.1% A, 34.3% T, 15.6% C and 16.0% G, with a bias towards AT (68.4%) and GC content of 31.6%.
In general, mitogenomic coding is associated with the mitochondrial translation apparatus, electron transport, and oxidative phosphorylation. The potential coding genes (PCGs) of Malassezia species included 14 PCGs related to oxidative phosphorylation [NAD dehydrogenase (nad1-6 and nad4L), cytochrome oxidases (cox1, cox2 and cox3), cytochrome b (cob), and 3 ATP synthases (atp6, atp8 and atp9), ribosomal protein S3 (rps3)] and 2 ribosomal RNA genes (rns and rnl). Only M. equina, M. caprae and M. vespertilionis presented 14 PCGs, due to the absence of the atp9 gene. This distribution is consistent with that observed in typical fungal mitogenomes, such as those belonging to Ascomycota and Basidiomycota (Dikarya Subkingdom). In total, the coding regions containing the 15 PCGs described occupied between 13,725 bp and 14,103 bp in the two mitogenomes presented (M. nana and M. dermatis, respectively). Likewise, the 15 PCGs occupied from 14,037 bp to 14,208 bp among the 6 new mitogenomes of M. pachydermatis described here, in addition to 12,735 bp and 14,784 bp among the mitogenomes available in the NCBI database. In mitogenomes that presented 14 PCGs, the coding regions occupied between 13,437 bp and 14,079 bp (M. equina and M. caprae, respectively). Other existing regions in mitogenomes are tRNAs, rRNA genes and intergenic regions. More details on these regions contained in mitogenomes are available in Supplementary Table 1.
Except for M. vespertilionis, all new mitogenomes showed nad4 and atp6 in the light chain. Likewise, a duplication of the atp9 gene in M. dermatis, M. nana and in the new M. pachydermatis mitogenomes was also observed in the light chain. In M. vespertilionis, only the cob gene was observed in the light chain. Apart from those already described, all other genes were observed in the heavy chain for all new mitogenomes. More details on the direction of PCGs can be found in Supplementary Tables 1-2.
Besides M. caprae and M. equina, where 18 tRNA genes were identified in the mitogenomes, the other species characterized in this work presented between 20 and 25 tRNAs, encoding the 20 standard amino acids. The length of tRNA genes in the five mitogenomes ranged from 71 bp to 86 bp. Each of the five Malassezia mitogenomes involved 2 rRNA genes: the small subunit ribosomal RNA gene (rns) and the large subunit ribosomal RNA gene (rnl). No significant difference was identified in the number of rRNA genes in the five species, and the total length of the genes.
Phylogenetic Analysis
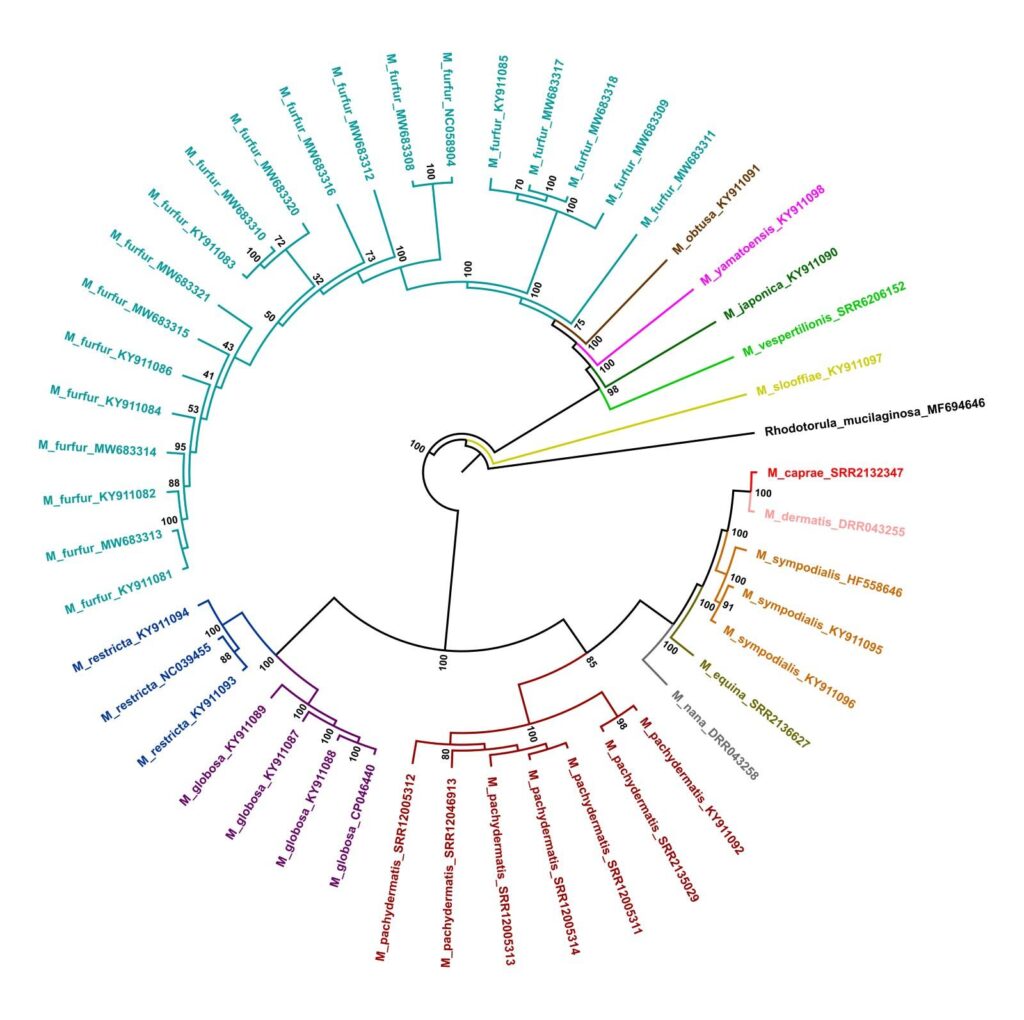
We obtained a robust evolutionary tree topology (Figure 1) with all recovered clades well supported. Our phylogeny is mostly accordant with a prior study based on sequences of six mitochondrial and nuclear genes (Wang et al., 2014). The tree has shown two main clades comprising a distinct group of species. As in the literature, our data reached a clade encompassing M. caprae, M. dermatis, M. sympodialis, M. equina, and M. nana. This clade is near related to M. pachydermatis clade. Another well-marked and related clade comprises M. globosa and M. restricta lineages.
The other main clade assembles M. sloffiae, M. vespertilionis, M. japonica, M. yamatoensis, M. obtusa, and a species complex formed by samples identified as M. furfur. Our analysis shows two clades of M. furfur with 100% bootstrap confidence and a single strain diverging early. The bootstrap confidence of this strain with the two largest clades reached 100% of confidence.
The main difference between our analysis and Wang et al. (2014) study was the position of M. restricta, M. globosa, and M. pachydermatis. In our three (Figure 1), these species are closest to the first mentioned clade, though previous analyzes grouped them with the second clade (Wang et al., 2014). In the phylogenetic analysis, the novel Malassezia pachydermatis mitogenome had clustered consistently with M. pachydermatis strain CBS1879 (KY911092.1). The same had happened with other species with multiple mitogenomes available.
Conclusion
The methodology used to assembly the mitochondrial genomes of Malassezia vespertilionis, Malassezia dermatis, Malassezia nana, Malassezia equina and Malassezia caprae proved to be satisfactory and allowed the expanded the phylogeny of Malasseziales through mtDNA. We were also able to validate our workflow by assembling 6 new Malassezia pachydermatis mitogenomes that demonstrated high agreement with the mitogenome already available. Furthermore, the study of the complete mitochondrial genome proves to be a tool with potential to solve taxonomic problems and help in understanding the evolutionary and ecological relationships of this group.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
RCO, IHRO, GH, GET, PST, RRR, DAB, DLJ, KFK, RP and FBM contributed to the conceptualization, investigation and design of the study. RCO, IHRO, GH, GET, PST, RRR, DAB, DLJ, KFK, RP and FBM performed the bioinformatics/statistics analysis and were responsible for data validation. FBM and DLJ provided resources to carry out the work. FBM, DLJ, KFK and RP were responsible for the administration and supervision of the research project. FBM, DAB, RCO, IHRO, PST and RRR wrote the first draft of the manuscript. FBM, DLJ, KFK and RP wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Acknowledgments
This study was financed in part by scholarship grants from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil – CAPES (www.capes.gov.br/) and Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (www.gov.br/cnpq/pt-br).
Supplementary Material
The Supplementary Material for this article can be found online at: https://doi.org/10.6084/m9.figshare.23666289.v1
Reference
Afgan E, Baker D, Batut B, Van Den Beek M, Bouvier D, Čech M et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic acids research. 2018;46(W1):W537-W544.
Al Arab M, Zu Siederdissen CH, Tout K, Sahyoun AH, Stadler PF, Bernt M. Accurate annotation of protein-coding genes in mitochondrial genomes. Molecular phylogenetics and evolution. 2017;106: 209-216.
Alikhan, NF., Petty, N.K., Ben Zakour, N.L. et al. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, 402 (2011). https://doi.org/10.1186/1471-2164-12-402
Aykut B, Pushalkar S, Chen R et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574:264–7.
Dierckxsens N, Mardulyn P, Smits G. NOVO Plasty: assembly of organelle genomes from whole genome data. Nucleic acids research. 2017;45(4):e18-e18.
Findley K, Oh J, Yang J et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2020;498:367–70.
Hirai A, Kano R, Makimura K, Duarte ER, Hamdan JS, Lachance MA, Yamaguchi H, HasegawaA. Malassezia nana sp. nov., a novel lipid-dependent yeast species isolated from animals. Int J Syst Evol Microbiol. 2004 Mar;54(Pt 2):623-627. doi: 10.1099/ijs.0.02776-0. PMID: 15023986.
Kalyaanamoorthy S, Minh BQ, Wong TK, Von Haeseler A, Jermiin LS. Model Finder: fast model selection for accurate phylogenetic estimates. Nature Methods 2017;4(6):587-589.
Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution. 2013;30(4):772– 780.
Limon JJ, Tang J, Li D et al. Malassezia is associated with Crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe. 2019;25:377–88.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, et al. IQTREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution 2020;37(5):1530-1534.
Saunte DML, Gaitanis G, Hay RJ. Malassezia-Associated skin diseases, the use of diagnostics and treatment. Front Cell Infect Microbiol. 2020;10:112.
Spatz M, Richard ML. Overview of the potential role of Malassezia in gut health and disease. Front Cell Infect Microbiol. 2020;10:201.
Theelen B, Cafarchia C, Gaitanis G et al. Malassezia ecology, pathophysiology, and treatment [published correction appears in Med Mycol. 2019 Apr 1;57(3):e2]. Med Mycol. 2018;56:S10– 25.
Vaidya G, Lohman DJ, Meier R. Sequence Matrix: concatenation software for the fast assembly of multi‐gene datasets with character set and codon information. Cladistics 2011;27(2):171-180.
Wang QM, Theelen B, Groenewald M, Bai FY, Boekhout T. Moniliellomycetes and Malasseziomycetes, two new classes in Ustilaginomycotina. Persoonia-Molecular Phylogeny and Evolution of Fungi. 2014;33(1):41-47.
Wu, G., Zhao, H., Li, C., Rajapakse, M. P., Wong, W. C. et al. Genus-wide comparative genomics of Malassezia delineates its phylogeny, physiology, and niche adaptation on human skin. PLoS Genetics 11, e1005614 (2015)
FIGURE
Figure 1.
1 Integrated Biotechnology Center, University of Mogi das Cruzes, Mogi das Cruzes, SP, Brazil
2 Technological Research Center, University of Mogi das Cruzes, Mogi das Cruzes, SP, Brazil
3 Laboratory of Ecological and Evolutionary
Genetics, Federal University of Viçosa, Rio Paranaíba, MG, Brazil.