CLINICAL MANIFESTATIONS AMONG PATIENTS WITH LEIGH SYNDROME IN BRAZIL
REGISTRO DOI: 10.5281/zenodo.8002247
Giovanna Maria de Freitas Oliveira1
Laércio Pol-Fachin2
RESUMO
INTRODUÇÃO: Síndrome de Leigh é uma doença rara e que faz parte das encefalopatias mitocondriais. Ela tem um caráter neurodegenerativo e tem como principal origem a genética. Sua principal forma é a infantil, com quadro clínico bastante variável. Associado à clínica, tem-se os exames laboratoriais, que juntos sugerem uma suspeita acerca da doença. OBJETIVO: Considerando a ausência de um quadro clínico mais detalhado e específico, o presente estudo teve por objetivo caracterizar as principais manifestações, sinais e sintomas da doença entre portadores no Brasil. METODOLOGIA: Para tal, realizou-se um estudo observacional analítico transversal, a partir da aplicação de um questionário entre os responsáveis pelos portadores da doença. RESULTADOS: Observou-se predominância estatisticamente significativa da doença em indivíduos de raça branca (p = 0.0017), com equivalência de ocorrência entre sexos. A maioria dos responsáveis percebeu que havia algo errado quando os portadores tinham entre um e dois anos, com predominância estatisticamente significativa de primeiros sintomas motores (p < 0.0001), mais associados a coordenação motora e tônus muscular, sem febre associada. Atualmente, os sistemas mais acometidos são o muscular, digestório, nervoso, respiratório e sensorial. CONSIDERAÇÕES FINAIS: Espera-se que os resultados possam contribuir para um diagnóstico mais precoce nesses pacientes, especialmente pois foram analisados quais os primeiros sintomas e quando eles costumam aparecer.
ABSTRACT
INTRODUCTION: Leigh syndrome is a rare disease that belongs to the group of mitochondrial encephalopathies. It has a neurodegenerative nature and its main origin is genetic. Its most common form is the infantile onset, with a highly variable clinical presentation. Laboratory tests are often used in conjunction with clinical findings to suggest a diagnosis. OBJECTIVE: Considering the absence of a detailed and specific clinical profile, the present study aimed to characterize the main manifestations, signs, and symptoms of the disease among carriers in Brazil. METHODOLOGY: An analytical cross-sectional observational study was conducted, using a questionnaire administered to caregivers of individuals with the disease. RESULTS: There was a statistically significant predominance of the disease in individuals of white race (p = 0.0017), with an equal occurrence between sexes. Most caregivers noticed that something was wrong when the affected individuals were between one and two years old, with a statistically significant predominance of initial motor symptoms (p < 0.0001), mostly related to motor coordination and muscle tone, without associated fever. Currently, the most affected systems are the muscular, digestive, nervous, respiratory, and sensory systems. CONCLUSIONS: It is hoped that the results of this study can contribute to earlier diagnosis in these patients, especially by identifying the first symptoms and their typical age of onset.
1 INTRODUÇÃO
A síndrome de Leigh (SL) é uma doença rara, neurometabólica congênita, que entra no grupo das encefalopatias mitocondriais [1]. É uma síndrome causada por déficit na produção de energia e que gera defeitos nos genes que são responsáveis pela codificação de alguns dos complexos mitocondriais [2]. Doença de caráter neurodegenerativo com acometimentos cognitivos e motores, levando à maioria das manifestações clínicas [2].
A SL tem origem genética em 50% dos casos, com diversas mutações identificadas no DNA mitocondrial ou nuclear, sendo as primeiras, responsáveis por 10 a 30% dos casos. O gene ATPase6 é o gene mitocondrial com maior prevalência de mutação dentre os doentes [3].
A clínica da SL é muito variável, pois não se enquadra no padrão dos achados clínicos das patologias mitocondriais, dificultando um diagnóstico precoce [3]. O quadro se inicia geralmente aos dois anos de idade, na maioria dos casos com uma evolução progressiva e insidiosa. Os sintomas da doença, em geral, podem surgir de duas formas: abrupta ou subaguda [1].
A forma mais comum da doença é a infantil [3], e o quadro clínico se diferencia pela faixa etária, entre até um ano e após um ano. Os primeiros sintomas da faixa etária inicial geralmente incluem: hipotonia, vômito, convulsão, irritabilidade, dificuldade de sucção. Já na segunda faixa etária, é mais comum a ataxia, dificuldade na marcha, disartria, regressão intelectual, distúrbios respiratórios e oftalmológicos [1].
Associado a clínica, tem-se os exames laboratoriais, que juntos sugerem uma suspeita acerca da doença. Aumento do lactato e piruvato sanguíneo, hiperproteinorraquia, proporção lactato/piruvato aumentada no sangue e líquor e hiperlactacidemia por sobrecarga glicídica, levam a suspeita da SL [1].
Após a forte suspeita da SL, se inicia uma investigação para confirmação da doença, e o seguinte exame solicitado é a ressonância magnética cranioencefálica, que pode encontrar em T2 as hiperintensidades focais, bilaterais e simétricas, mais comumente na região dos gânglios da base ou tronco cerebral. Esses achados também podem ser encontrados no anatomopatológico e são concedidos à depleção do ATP, levando a lactoacidose, seguido da congestão vascular e hipóxia, resultando na necrose [3].
Até o momento, não existe um tratamento específico e comprovado. Atualmente, é feito o acompanhamento de toda a equipe de saúde para cada necessidade, e um cuidado paliativo com o portador da síndrome. O prognóstico é relativo para cada caso e cada paciente [3].
Devido a SL ser uma patologia com um amplo quadro clínico, observa-se uma maior dificuldade, levando aos médicos recorrerem a tratamentos comuns, tais como fisioterapia, fonoaudiologia, um tratamento multidisciplinar, pois não se sabe o diagnóstico correto [3].
Há uma escassez de estudos a respeito da SL na literatura, sem muitos trabalhos bem definidos sobre sua fisiopatologia, epidemiologia, clínica, diagnóstico e tratamento. Nesse sentido, por não se ter muito conhecimento sobre essa doença, esse trabalho pretende abranger um lado mais clínico da doença, fazendo uma busca sobre a prevalência das patologias associadas a essa síndrome no Brasil.
2 METODOLOGIA
Trata-se de um estudo observacional, analítico e transversal com abordagem quali-quantitativa. A pesquisa foi desenvolvida através de um questionário, em um grupo de WhatsApp com responsáveis dos portadores da síndrome de Leigh, realizado no mês de novembro de 2022.
A amostra foi composta por 20 portadores da síndrome de Leigh. O recrutamento foi iniciado com uma campanha no grupo do Whatsapp, passando as informações sobre a doença e explicando a necessidade de consentimento voluntário e aplicação do Termo de Consentimento Livre e Esclarecido (TCLE). Vale ressaltar que foram os responsáveis pelos portadores da síndrome que colaboraram com a pesquisa. O grupo que fez parte da pesquisa, em si, já é um grupo vulnerável, pois são pessoas, que na maioria dos casos, não respondem por si mesma, necessitando de um responsável. E esse grupo, é de extrema importância para um maior conhecimento da síndrome. Essa pesquisa foi aprovada por um comitê de ética em pesquisa, sob CAAE 60689822.5.0000.0039 e número do parecer de aprovação 5.706.270.
Após o consentimento para participação no estudo, foi disponibilizado o questionário de pesquisa, contendo perguntas sobre a clínica dos portadores da síndrome, expondo sistema a sistema, para que marcassem quais sistemas estão acometidos pelo portador e mais adiante, identificar qual o déficit específico. Foi coletado a idade atual do portador, a idade em que foi diagnosticada a doença, qual o primeiro sintoma que o levou a buscar ajuda médica, qual o primeiro sistema acometido, e quais os atuais sistemas acometidos, tudo isso foi feito em uma linguagem mais informal para que as pessoas leigas pudessem compreender bem.
Após a coleta dos dados, já tabulados em planilhas eletrônicas, a análise dos dados quantitativos foi feita por meio de estatística descritiva, através do cálculo de frequências absolutas e relativas para as variáveis estudadas. Foi também aplicada estatística analítica, nas quais foram consideradas significativas as análises os valores de p menores que 0,05, através dos testes de qui-quadrado de uma e duas via, para avaliar as diferenças observadas dentro de cada variável e a associação entre variáveis qualitativas dicotômicas. Os dados qualitativos foram analisados por meio da análise de conteúdo. Para garantir o anonimato, os depoimentos dos participantes foram representados pela letra E maiúscula significando entrevistado, acrescida do numeral correspondente à ordem de recebimento das respostas no Google Forms, como por exemplo, E1, E2 e assim sucessivamente.
3 RESULTADOS
Os vinte responsáveis que participaram da pesquisa afirmaram que os portadores de SL possuem entre 3 e 27 anos, com média de 11 e mediana de 8,5. A medida de tempo de convivência com a doença é de 7,9 anos, com mediana de 6 anos, e tempo máximo de até 26 anos. Observou-se predominância estatisticamente significativa de indivíduos de raça branca (qui-quadrado de uma via, p = 0.0017), e equivalência de ocorrência entre sexos, com discreta predominância entre mulheres (Tabela 1).
Tabela 1. Dados sociodemográficos e outras características dos portadores de SL.
Categorias Frequência % p-valor* Sexo Feminino 11 55 0.6547 Masculino 9 45 Raça Branca 17 85 0.0017 Parda 3 15 Faixa etária Até 6 anos 6 30 0.3080 De 7 a 12 anos 8 40 De 13 a 18 anos 3 15 Maiores de 18 anos 3 92
* Teste de qui-quadrado de uma via (One-Way Chi-Squared test)
Com relação ao surgimento da doença, observou-se que a maioria dos responsáveis percebeu que havia algo errado quando os portadores tinham entre um e dois anos, mesma idade no qual o diagnóstico foi confirmado para a maioria dos portadores (Tabela 2). De acordo com os dados obtidos, observou-se predominância estatisticamente significativa de primeiros sintomas motores (qui-quadrado de uma via, p < 0.0001), e a maioria dos participantes não apresentou febre junto com esses primeiros sinais (Tabela 2). Não foi observada associação estatisticamente significativa na busca por associações entre ocorrência de febre, etnia e sexo.
Dentre os relatos de qual foi o primeiro sintoma que levou à procura de ajuda médica, houve uma grande variabilidade de respostas. No entanto, as mais recorrentes foram dificuldades associadas a coordenação motora e tônus muscular (12), atraso no desenvolvimento (4), e nistagmo (2).
Tabela 2. Dados a respeito do surgimento da SL entre os participantes
Categorias Frequência % p-valor* Idade em que se percebeu que algo estava errado Menos de um ano 6 30 0.2466 Entre um e dois anos 10 50 Mais de dois anos 4 20 Idade do portador quando SL foi diagnosticada Menos de um ano 2 10 0.1577 Entre um e dois anos 9 45 Entre três e quatro anos 4 20 Mais de quatro anos 5 25 Febre junto aos primeiros sintomas Sim 7 35 0.1797 Não 13 65 Primeiro sistema acometido§ Muscular 15 75 < 0.0001 Imunológico 3 15 Nervoso 3 15 Sensorial 3 15 Digestório 2 10 Urinário 2 10
* Teste de qui-quadrado de uma via (One-Way Chi-Squared test)
§ Os valores, somados, superam os 100% pois algumas respostas incluíam múltiplos sistemas
Com relação aos sistemas acometidos nos pacientes atualmente, houve relatos de mais que o dobro de sistemas apresentando sintomas, em comparação com os primeiros observados (Figura 1). No entanto, o sistema muscular segue sendo o mais afetado, mas com grande prevalência também dos sistemas digestório, nervoso, respiratório, sensorial e imunológico (Figura 1).
Figura 1. Sistemas acometidos atualmente em pacientes portadores da SL, 2023
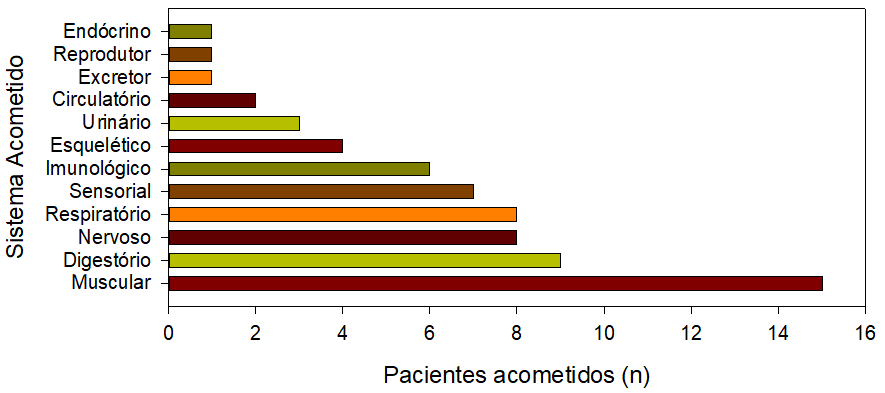
Quando questionados sobre os problemas apresentados pelo portador, seguindo os dados observados na Figura 1, a maioria dos relatos apontam para problemas de ordem muscular, tal como descritos abaixo:
Devido ao prejuízo de tônus muscular e desordens metabólicas, quanto a metabolização de energia pelas células, há um prejuízo neurológico que ocasiona grande espasticidade de membros inferiores e superiores e hipotonia muscular. Essa hipotonia favoreceu o desenvolvimento de uma escoliose, a espasticidade de membros inferiores favoreceu a formação de uma luxação de quadril, e flexão de membros superiores com perda funcional gradativa de mobilidade e funcionalidade de mãos e pés (E11).
Hoje ela é cadeirante, tem uma distonia generalizada muito grave, não se senta, não faz trocas posturais eficazes, tem instabilidade no controle cervical, não fala, come por boca, mas não tem uma mastigação boa, tem nistagmo, tem atrofia no nervo óptico. Sabemos que enxerga, porque responde por figuras quando quer, se comunica bem através de gestos e expressões faciais. Percebemos que tem uma boa compreensão do que é falado pra ela, é super participativa nas terapias, mas a dificuldade motora é muito grande mesmo. A distonia judia demais dela, ela faz uso de botox para aliviar (E20).
Da mesma forma, vários relatos apontam dificuldades no sistema digestório, nas quais relatos de engasgo, dificuldades na mastigação e deglutição são os mais comuns:
Não anda, se engasga muito e sua imunidade é muito baixa (E2).
A coordenação motora de membros superiores está atrofiada, não anda, não fala, comportamento infantil, usa sonda de gastrostomia endoscópica percutânea (gtt) para a alimentação, usa fralda e cadeirante tem 23 anos de idade (E5).
Outros relatos apontam para o acometimento respiratório e sensorial, nas quais foram bastante prevalentes relatos de surdez, ausência de fala e cegueira:
Ele é acamado dependendo do ventilador para respirar, não fala, não anda, não enxerga (E10).
Estrabismo nos olhos, um pouco de paralisia facial (E18).
4 DISCUSSÃO
O primeiro caso da SL no Brasil descrito na literatura foi de uma menina diagnosticada aos 10 anos de idade [1], na qual foi observado que as manifestações clínicas são variadas e se iniciam geralmente até os 2 anos de idade. Há relatos de crianças cujo diagnóstico e óbito ocorreram antes de um ano de idade [4]. Comparando com o que foi coletado na pesquisa, observa-se que existe uma predominância de ocorrência em indivíduos caucasianos, tal como em casos relatados a nível nacional [1,4], que existe uma discreta prevalência em meninas, e que a maioria dos sintomas iniciais foram percebidos até os dois anos de idade. Ainda, embora existam uma variedade de sintomas iniciais, a literatura indica que lactato elevado (denotando problemas musculares) e atrasos no desenvolvimento são alguns dos mais comuns [5]. Nesse trabalho, mostrou-se que existe uma predominância de acometimento no sistema muscular, seguido do digestório.
Observa-se também que a evolução é insidiosa e progressiva que já havia sido referida em outra literatura, pôde ser confirmada já que existe portadores que já estão com mais de 18 anos. Pelo caráter progressivo da doença, foi possível analisar a evolução da clínica desses pacientes, com um aumento significativo do número de sistemas acometidos ao longo do tempo. Dessa forma, observa-se o quanto a doença tem um aspecto debilitante. A febre, também como já havia sido mencionada em outros artigos, foi constatada que existe uma relação com a doença, pois 35% dos portadores estudados iniciaram com quadro de febre junto aos primeiros acometimentos.
5 CONCLUSÃO
Os dados coletados apontam para uma maior prevalência de sintomas musculares entre os acometidos pela doença no Brasil, incluindo fraqueza ou perda de movimentos corporais. Não obstante, existem relatos associados a problemas em mais de dez sistemas fisiológicos, incluindo digestório (dificuldades na mastigação e deglutição), e sensorial (cegueira, surdez e ausência de fala). Observou-se uma maior prevalência em indivíduos de etnia branca, com acometimento equivalente entre meninos e meninas.
Espera-se que a pesquisa contribua para a melhoria dos estudos acerca da doença, permitindo contribuir para o entendimento dos principais sistemas acometidos, a fim de direcionar medidas que minimizem os sintomas e a progressão da doença, melhorando a qualidade de vida tanto dos pacientes quanto dos seus responsáveis e cuidadores.
REFERÊNCIAS
- Roma AC, Pereira PRAA, Dantas, AM. Síndrome de Leigh: relato de caso. Arquivos Brasileiros de Oftalmologia. v. 71, n. 1, p. 118-121, 2008.
- Herrera, MD, Garcia EE, Morales JMG, Pena OYE. Diagnóstico clínico e bioquímico da síndrome de Leigh em cinco pacientes colombianos. Revista Med, v. 26, n. 1, p. 26-33, 2018.
- Lopes T, Coelho M, Bordalo D, Bandeira A, Bandeira A, Vilarinho L, Fonseca P, Carvalho S, Martins C, Olivera JG. Síndrome de Leigh: a propósito de um caso clínico com mutação no DNA mitocondrial. Revista Paulista de Pediatria, v. 36, n. 4, p. 519-523. 2018.
- Tormen M, Nunes TM, Dal`Bó K. Síndrome de Leigh. Revista Brasileira de Neurologia e Psiquiatria. V 20, n. 3, p. 267-271, 2016.
- Stenton SL, Zou Y, Cheng H, Liu Z, Wang J, Shen D, Jin H, Ding C, Tang X, Sun S, Han H, Ma Y, Zhang W, Jin R, Wang H, Sun D, Lv JL, Prokisch H, Fang F. Leigh Syndrome: A Study of 209 Patients at the Beijing Children’s Hospital. Annals of Neurology, v. 91, n. 4, p. 466-482, 2022.
1Giovanna Maria de Freitas Oliveira Acadêmica de Medicina do Centro Universitário CESMAC Rua Cônego Machado, 918 – Farol, Maceió – AL CEP: 57051-160 giovannafreitaso@hotmail.com
2Laércio Pol-Fachin Doutor em Biologia Celular e Molecular Centro Universitário CESMAC Rua Cônego Machado, 918 – Farol, Maceió – AL CEP: 57051-160 laercio.fachin@cesmac.edu.br