REGISTRO DOI: 10.5281/zenodo.7695928
Estevão Moreira David
Rodrigo Bertani Simão
Siliyizeth Gómez Restrepo
Carlos Vaz de Melo Maciel
Otávio Augusto Fonseca Reis
Relato de caso
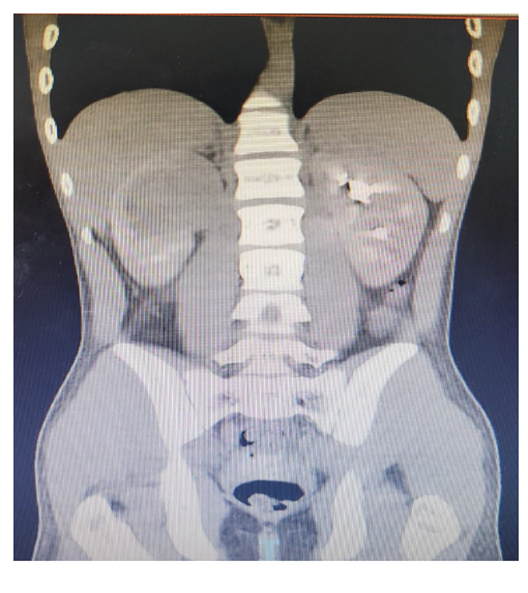
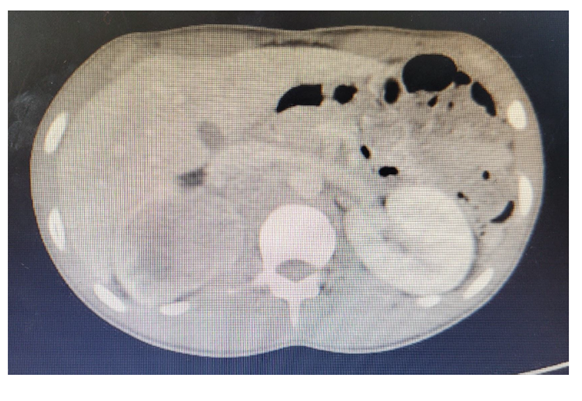
Paciente do sexo masculino, 17 anos, melanoderma, atendido no pronto atendimento do Hospital Felício Rocho com relato de dor contínua em flanco direito há 1 semana, sem sintomas urinários. Nega febre. Relato de hematúria macroscópica há 2 meses com resolução espontânea. Nega comorbidades ou cirurgias prévias. Exame físico e exames laboratoriais sem alterações significativas. A tomografia computadorizada (Figura 1) mostrava rim direito com massa heterogênea e hipoatenuante centrada no polo superior, de limites imprecisos, com focos de liquefação/necrose, associando-se linfonodomegalia retroperitoneal. Realizada biópsia percutânea guiada por ultrassom para diagnóstico histológico da lesão. Anatomopatológico revelou se tratar de um carcinoma medular renal.
Optou-se por realizar nefrectomia radical direita videolaparoscópica com linfadenectomia retroperitoneal, transcorrendo o procedimento cirúrgico sem intercorrências. Durante o ato operatório foi identificado acometimento de linfonodos interaórtico cavais irressecáveis. A evolução pós-operatória imediata foi satisfatória.
Exame anatomopatológico compatível com carcinoma medular renal, medindo 6 cm no maior eixo com invasão da gordura hilar. Presença de metástase em 1 de 2 linfonodos dissecados. O estadiamento final (TNM) foi pT3a pN1 pMX. Paciente encaminhado para seguimento oncológico.
Figura 1 – Tomografia computadorizada
Discussão
O carcinoma medular renal (CMR) é uma neoplasia altamente agressiva, descrita pela primeira vez por Davis em 1995, como a ‘’sétima nefropatia falciforme’’. Acomete principalmente pacientes jovens de ascendência africana com traço falciforme. No entanto, também pode ocorrer em hispânicos e caucasianos, particularmente naqueles de ascendência do sul da Europa.¹
Trata- se de tumor raro, sendo responsável por < 0,5% dos carcinomas renais, sua prevalência pode ser subestimada devido à dificuldade em diferenciá-lo do carcinoma do ducto coletor (CDC) e outras malignidades renais. A maioria dos pacientes tem entre 20-30 anos, com predominância do sexo masculino e predileção pelo rim direito (3:1). Seu tamanho varia de 2 a 18 cm, com diâmetro médio de 7,4 cm.²,³,⁴
A patogênese da CMR não é completamente compreendida. Supõe-se que esteja relacionado à hipóxia medular crônica. Postula-se que a hipóxia e a hiperosmolaridade extremas na medula renal, bem como a isquemia regional induzida pela falcização das hemácias, ativem mecanismos de reparo de DNA que levam a deleções e translocações em SMARCB1.
SMARCB1 é um gene supressor de tumor do cromossomo 22 na posição 11.23 (22q11.23), funcionando como parte do regulador dependente de actina do complexo de remodelação da cromatina, que é importante na regulação gênica. O diagnóstico do CMR é confirmado pela demonstração da perda de coloração nuclear SMARCB1 (INI1).⁴,⁵,⁶
O tumor apresenta-se como uma massa mal circunscrita na região medular, comumente apresentando quantidades variáveis de hemorragia e necrose. Frequentemente apresentam extensão extrarrenal no momento da apresentação. Pequenos nódulos satélites são comumente encontrados no córtex renal. Microscopicamente, observa-se um padrão reticular ou cribriforme característico com uma resposta estromal desmoplásica marcante e um infiltrado inflamatório misto robusto.²,⁴
Radiograficamente, são tumores centrais e demonstram um padrão infiltrativo, distintamente diferente da aparência das massas periféricas exofíticas de CCR de células claras.⁷
O diagnóstico diferencial deve ser feito com o carcinoma de ducto coletor, o tumor rabdóide maligno, o carcinoma urotelial e com os outros subtipos de carcinoma de células renais.²
A maioria dos pacientes (44%) apresenta hematúria e/ou dor no flanco (44%) como sintomas iniciais. Perda de peso (26%), desconforto respiratório (14%), massa palpável, tosse e febre também são relatados. A grande maioria dos pacientes apresenta doença metastática ao diagnóstico, sendo que 87% tem metástase linfonodal. O CMR geralmente tem um curso rapidamente progressivo. Além da disseminação linfovascular é comum que haja invasão direta das veias do seio renal e do retroperitônio.³,⁴
As metástases à distância são relatadas principalmente para glândulas adrenais, pulmão, fígado, couro cabeludo, cérebro ou órbita.⁸
O CMR é resistente a todas as terapias direcionadas comumente usadas contra outros carcinomas de células renais e é altamente agressivo, com < 5% dos pacientes sobrevivendo por mais de 36 meses. Não há tratamento padrão para o seu gerenciamento e modelos foram extrapolados de protocolos para carcinoma de células uroteliais ou tumores renais primários. 9,10
Para o CMR localizado, que se apresenta em apenas 5% dos casos, o recomendado é a nefrectomia associada a algum tipo de linfadenectomia, seguida de monitoramento rigoroso ou associação com algum tipo de terapia sistêmica ou quimioterapia. O benefício da nefrectomia radical não é estatisticamente significativo em pacientes com doença metastática.³,⁹
Vários esquemas de terapia sistêmica são descritos: (a) gencitabina/bevacizumab/paclitaxel; (b) MVAC (metotrexato, vinblastina, Adriamicina e cisplatina); (c) sunitinibe/ everolimus ou doxorrubicina; (d) etoposido/ifosfamida; (e) bevacizumabe/everolimus; (f) monoterapia com nivolumabe; (g) monoterapia com ipilimumabe; e (h) bortezomibe/ pazopanibe. Na maioria dos casos ocorre uma resposta parcial seguida de progressão rápida da doença.³,⁹
O CMR é um câncer altamente agressivo, com má resposta ao tratamento, com sobrevida média de 4 a 5 meses nos casos com doença metastática no momento da apresentação e média de 17 meses em pacientes sem doença metastática.³,⁸
Metástase linfonodal, a não realização de nefrectomia e um ECOG PS de 2-3 na apresentação foram associados a pior sobrevida.⁴
Os locais mais comuns de recidiva são pulmão, pleura, linfonodos mediastinais e osso.¹¹
O CMR é uma doença rara, que se apresenta principalmente em homens jovens, de origem africana com traço falciforme. É altamente agressivo, e a maioria dos pacientes apresenta metástases ao diagnóstico. Apresenta opções de tratamento limitadas e desfechos universalmente ruins. Os pacientes que foram submetidos à nefrectomia durante o curso da doença tiveram uma sobrevida maior do que aqueles que foram tratados apenas com terapia sistêmica. Estudos são urgentemente necessários para identificar alterações moleculares que possam fornecer a base para o desenvolvimento de uma terapia nova, racional e mais eficaz para o CMR.
Referências
Davis Jr, Charles J., F. K. Mostofi, and Isabell A. Sesterhenn. “Renal medullary carcinoma. The seventh sickle cell nephropathy.” The American journal of surgical pathology 19.1 (1995): 1-11.
Elliott, Alexis, and Evelyn Bruner. “Renal medullary carcinoma.” Archives of Pathology & Laboratory Medicine 143.12 (2019): 1556-1561.
Blas, Leandro, et al. “Renal medullary carcinoma: a report of the current literature.” Current Urology Reports 20.1 (2019): 1-8.
Shah, Amishi Y., et al. “Management and outcomes of patients with renal medullary carcinoma: a multicentre collaborative study.” BJU international 120.6 (2017): 782-792.
Calderaro, Julien, et al. “Balanced translocations disrupting SMARCB1 are hallmark recurrent genetic alterations in renal medullary carcinomas.” European urology 69.6 (2016): 1055-1061.
Msaouel, Pavlos, Nizar M. Tannir, and Cheryl Lyn Walker. “A Model Linking Sickle Cell Hemoglobinopathies and SMARCB1 Loss in Renal Medullary CarcinomaPathogenesis of Renal Medullary Carcinoma.” Clinical Cancer Research 24.9 (2018): 2044-2049.
Greco, Federico, et al. “Imaging of renal medullary carcinoma.” Journal of Kidney Cancer and VHL 4.1 (2017): 1.
Su, Yongdong, and Andrew L. Hong. “Recent Advances in Renal Medullary Carcinoma.” International Journal of Molecular Sciences 23.13 (2022): 7097.
Iacovelli, Roberto, et al. “Clinical outcome and prognostic factors in renal medullary carcinoma: a pooled analysis from 18 years of medical literature.” Canadian Urological Association Journal 9.3-4 (2015): E172.
Msaouel, Pavlos, et al. “Updated recommendations on the diagnosis, management, and clinical trial eligibility criteria for patients with renal medullary carcinoma.” Clinical genitourinary cancer 17.1 (2019): 1-6.
Amin, Mahul B., et al. “Collecting duct carcinoma versus renal medullary carcinoma: an appeal for nosologic and biological clarity.” The American journal of surgical pathology 38.7 (2014): 871-874.
¹Davis Jr, Charles J., F. K. Mostofi, and Isabell A. Sesterhenn. “Renal medullary carcinoma. The seventh sickle cell nephropathy.” The American journal of surgical pathology 19.1 (1995): 1-11.
²Elliott, Alexis, and Evelyn Bruner. “Renal medullary carcinoma.” Archives of Pathology & Laboratory Medicine 143.12 (2019): 1556-1561.
³Blas, Leandro, et al. “Renal medullary carcinoma: a report of the current literature.” Current Urology Reports 20.1 (2019): 1-8.
⁴Shah, Amishi Y., et al. “Management and outcomes of patients with renal medullary carcinoma: a multicentre collaborative study.” BJU international 120.6 (2017): 782-792.
⁵Calderaro, Julien, et al. “Balanced translocations disrupting SMARCB1 are hallmark recurrent genetic alterations in renal medullary carcinomas.” European urology 69.6 (2016): 1055-1061.
⁶Msaouel, Pavlos, Nizar M. Tannir, and Cheryl Lyn Walker. “A Model Linking Sickle Cell Hemoglobinopathies and SMARCB1 Loss in Renal Medullary CarcinomaPathogenesis of Renal Medullary Carcinoma.” Clinical Cancer Research 24.9 (2018): 2044-2049.
⁷Greco, Federico, et al. “Imaging of renal medullary carcinoma.” Journal of Kidney Cancer and VHL 4.1 (2017): 1.
⁸Su, Yongdong, and Andrew L. Hong. “Recent Advances in Renal Medullary Carcinoma.” International Journal of Molecular Sciences 23.13 (2022): 7097.
⁹Iacovelli, Roberto, et al. “Clinical outcome and prognostic factors in renal medullary carcinoma: a pooled analysis from 18 years of medical literature.” Canadian Urological Association Journal 9.3-4 (2015): E172.
¹⁰Msaouel, Pavlos, et al. “Updated recommendations on the diagnosis, management, and clinical trial eligibility criteria for patients with renal medullary carcinoma.” Clinical genitourinary cancer 17.1 (2019): 1-6.
¹¹Amin, Mahul B., et al. “Collecting duct carcinoma versus renal medullary carcinoma: an appeal for nosologic and biological clarity.” The American journal of surgical pathology 38.7 (2014): 871-874.