REGISTRO DOI: 10.5281/zenodo.7384453
Eduardo Araújo Pereira
Orientador: Leonardo Guimarães de Andrade
RESUMO
A anemia falciforme é uma doença genética e hereditária, predominante em negros mais também pode manifestar-se em brancos. Ela também é caracterizada por uma deformação nos glóbulos vermelhos, que perdem a forma discoidal e bincôncova, adquirindo um aspecto de foice que endurecem e dificulta a passagem do sangue pelos vasos de pequeno calibre e a oxigenação dos tecidos. As hemácias falciformes contêm a hemoglobina S, que se cristalizam com a falta de oxigênio, formando trombos que dificultam a passagem do fluxo sanguíneo, porque não tem a maleabilidade da hemácia normal. A anemia falciforme é causada por uma mutação genética, responsável pela deformidade dos glóbulos vermelhos (hemácias). Para ser portador da anemia falciforme é preciso que o gene seja transmitido pela mãe ou pelo pai. Se o gene alterado for transmitido apenas por um dos pais, o filho terá traço falciforme, a doença não se manifesta, mas poderá passar para seus descendentes. O diagnóstico laboratorial é feito através da eletroforese de hemoglobina que é um exame específico para o diagnóstico da anemia falciforme, mas a hemoglobina S pode ser detectada pelo teste do pezinho quando a criança nasce.
Palavras-chave: Anemia Falciforme; Hemoglobinopatias; Diagnóstico Laboratorial.
ABSTRACT
Sickle cell anemia is a genetic and hereditary disease, predominant in blacks and may also manifest itself in whites. It is also characterized by a deformation in the red blood cells, which lose the discoid and biconcave form, acquiring an aspect of sickle that harden and makes difficult the passage of the blood by the vessels of small caliber and the oxygenation of the tissues. Sickle red cells contain hemoglobin S, which crystallize with lack of oxygen, forming thrombi that hinder the passage of blood flow because it does not have the malleability of the normal red blood cell. Sickle cell anemia is caused by a genetic mutation, responsible for deformity of red blood cells (red blood cells). To be a carrier of sickle cell anemia, the gene must be transmitted by the mother or the father. If the altered gene is transmitted only by the parents, the child will have a sickle cell trait, the disease will not manifest but may pass to his offspring. The laboratory diagnosis is made by hemoglobin electrophoresis, which is a specific test for the diagnosis of sickle cell anemia, but hemoglobin S can be detected by the test of the foot when the child is born.
Keywords: Sickle cell anemia; hemoglobinopathies; laboratory diagnosis
1 OBJETIVO GERAL
Descrever sobre a anemia falciforme e suas características, como a transmissão, sintomas, diagnóstico laboratorial no SUS e seu devido tratamento.
1.1 OBJETIVOS ESPECÍFICOS
-Descrever sobre a anemia falciforme;
-Apresentar os principais métodos de diagnóstico laboratorial da anemia falciforme no SUS;
-Esclarecer sobre as principais formas de tratamento.
2 JUSTIFICATIVA
É importante conhecer a anemia falciforme, porque ela está muito presente na nossa sociedade. Tendo a necessidade de um diagnóstico precoce para o tratamento adequado e acompanhamento ambulatorial específico. O diagnóstico precoce e o acompanhamento com equipe de profissionais treinados para o tratamento da anemia falciforme podem reduzir muito as complicações da doença e até ajudar a diminuir a taxa de mortalidade dos pacientes.
3 METODOLOGIA
O presente trabalho baseou-se em pesquisas bibliográficas realizadas entra o período de agosto e novembro de 2022, que assim possibilitaram o levantamento das informações necessárias para alcançar os objetivos do trabalho no qual as informações foram obtidas de Anvisa e artigos científicos a partir de fontes como, Scientific Eletronic Library Online (Scielo). Foram utilizadas palavras-chave: Anemia Falciforme. Hemoglobinopatias. Após a escolha dos artigos científicos, iniciou-se a análise dos mesmos a partir de uma leitura rigorosa para o reconhecimento dos artigos que fundamentariam a pesquisa.
4 INTRODUÇÃO
A anemia falciforme é uma enfermidade genética que afeta principalmente a população negra; é caracterizada pela presença de células vermelhas com formato anormal (forma de foice), que são removidas da circulação e destruídas. A alteração de base nas células vermelhas é a presença de uma hemoglobina anormal (HbS), que quando desoxigenada, se torna relativamente insolúvel, formando agregados que distorcem sua forma e impedem seu fluxo no interior dos vasos sangüíneos.
A maior prevalência da hemoglobina S (HbS) ocorre na África tropical e entre os negros nos países que participaram do tráfico de escravos. A presença da hemoglobina S em homozigose, num indivíduo caracteriza a anemia Falciforme, que é uma das doenças hereditárias mais comuns no Brasil, devido a miscigenação. A vinda de escravos para o Brasil possivelmente trouxe indivíduos do continente africano com a mutação genética produtora de HbS, onde houve a proliferação na população (SOUZA; 2009).
Por ser uma anemia hereditária comum no mundo, o reconhecimento tardio pode levar à morte nos primeiros anos de vida, uma vez que as internações concentram-se em faixas etárias jovens, observação condizente com a literatura, que revela o grande impacto social da doença e alerta quanto à importância de se aperfeiçoar a atenção aos pacientes portadores do gene falciforme (LOUREIRO; ROZENFELD; 2005).
DESENVOLVIMENTO
4.1 DESCRIÇÕES DA ANEMIA FALCIFORME
4.1.1 Histórico
A Anemia Falciforme (AF) foi apresentada na literatura pela primeira vez, pelo médico James Bryan Herrick, em 1910, que acompanhou um jovem negro das Antilhas emigrado para Chicago durante seis anos. O paciente apresentava um quadro clínico de aumento cardíaco, albuminúria, adenopatia, icterícia, dor nas articulações e anemia secundária não notável para a grande redução das hemácias ou hemoglobina (Hb), mas que se caracterizava pela forma atípica das hemácias, com tendência a assumir a forma de foice (HERRICK, 1910).
4.1.2 Bases teórico-conceituais da Doença Falciforme
Estudos antropológicos associados às análises biomoleculares sugerem que o gene anormal para a síntese da Hb S pode ter ocorrido há aproximadamente 50 a 100 mil anos, nas regiões centro-oeste da África, Índia e leste da Ásia. A denominação “anemia falciforme” é reservada para a forma da doença que ocorre nesses homozigotos SS. Além disso, o gene da HbS pode combinar-se com outras anormalidades hereditárias das hemoglobinas, como hemoglobina C (HbC), hemoglobina D (HbD), beta-talassemia, entre outros, gerando combinações que também são sintomáticas, denominadas, respectivamente, hemoglobinopatia SC, hemoglobinopatia SD, S/beta-talassemia. No conjunto, todas essas formas sintomáticas do gene da HbS, em homozigose ou em combinação, são conhecidas como “doenças falciformes”. As síndromes falciformes incluem ainda o traço falciforme (HbAS) e a doença falciforme associada à persistência hereditária de hemoglobina fetal (HbS/PHHF) (ANVISA, 2002).
4.1.3 Hemoglobina normal (Hb A)
O transporte de oxigênio para todos os tecidos humanos é feito através de pigmentos respiratórios denominados hemoglobina, que está localizada no interior das hemácias. A hemoglobina é composta pela conjugação de um pigmento, o heme, e de uma proteína, a globina. O heme é um complexo formado por um átomo de ferro, situado no interior de uma estrutura porfirínica que mantém o estado ferroso e dá a cor vermelha característica da hemoglobina. A globina consiste de dois pares de cadeias polipeptídicas – o tetrâmero – com um total de 574 aminoácidos formando uma molécula tetramerizada. O tetrâmero é constituído por duas cadeias globínicas do tipo alfa (α) que possuem 141 aminoácidos e cujos genes estão localizados no cromossomo 16 e por duas cadeias globínicas do tipo beta (β) com 146 aminoácidos localizados no cromossomo 11 (NAOUM, 1987).
4.1.4 Hemoglobinas anormais (Hb S)
Na DF há uma alteração na hemoglobina A (HbA), originando uma hemoglobina anormal denominada HbS (derivado do inglês sickle), por promover na hemácia desoxigenada mudança da forma bicôncava para a de foice, originando o termo “doença falciforme” ou “sickle cell disease” (LOBO; MARRA; SILVA, 2007). A HbS difere da HbA, figura1, tanto no aspecto estrutural quanto elétrico. Por ser uma anomalia da globina beta, as características clínicas dessa doença serão percebidas após a estabilização da produção das globinas, o que ocorre por volta do sexto mês de vida, quando a síntese da globina gama (Ɣ) (atuante na fase fetal) é interrompida, enquanto que o gene beta sintetiza em sua plenitude globinas beta normais ou anormais (NAOUM, 2000). As células falciformes têm sobrevida muito curta, de 16 a 20 dias, quando comparadas aos 120 dias da hemácia normal (ANVISA, 2002; NAOUM; NAOUM, 2004).
FIGURA 1: Exemplo de hemácias normais e hemácias falciformes
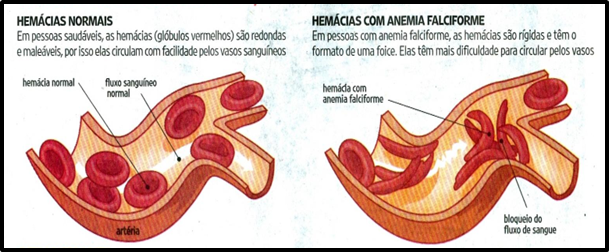
Fonte: ROCHA, W, 2022.
Na DF, em decorrência de uma mutação no cromossomo 11, no gene da globina beta S, ocorre a substituição da base nitrogenada adenina (A), do códon normal GAG pela timina (T) do códon GTG, resultando na substituição do sexto aminoácido da globina beta – o ácido glutâmico (GAG), por outro aminoácido diferente, a valina (GTG) (NAOUM, 2000; GALIZA NETO; PITOMBEIRA, 2003; LOBO; MARRA; SILVA, 2007).
4.1.5 Epidemiologia da doença
Atualmente, estima-se que existam no Brasil entre 20 a 30 mil portadores da doença falciforme sendo considerada pela coordenação da PNAIPDF do Ministério da Saúde, como um problema que requer medidas de saúde pública (BRASIL, 2009).
• 137.767 crianças com o traço falciforme (1:30).
• 2.934 crianças diagnosticadas com a doença falciforme, cerca de 180 casos por ano.
• 2.601 crianças com doença falciforme em acompanhamento ambulatorial.
• 1/1.400 é a incidência de crianças com doença falciforme. Isso equivale a cerca de 70 casos a cada 100 mil nascidos vivos (CEHMOB-MG, 2014).
4.2 FORMAS DE DIAGNÓSTICOS LABORATORIAIS DA ANEMIA FALCIFORME NO SISTEMA ÚNICO DE SAÚDE
As técnicas utilizadas para avaliar amostras de sangue com suspeitas de doença falciforme encaminhadas ao laboratório incluem testes que podem ser subdivididos em quatro grupos:
Cada grupo de testes tem sua importância na informação de dados para fundamentar com segurança o diagnóstico completo da doença Falciforme (NAOUM, PC; 2012)
4.2.1 Teste do Pezinho
O teste do pezinho é um exame de prevenção fundamental para a saúde da criança, pois garante que doenças raras sejam detectadas precocemente e o tratamento adequado iniciado o quanto antes. Por esse motivo, o procedimento deve ser realizado em um período específico: a partir do terceiro dia de vida do bebê e até no máximo no quinto dia após o nascimento (HADACHI S; 2017).
É importante que ele seja feito o mais rápido possível porque, com o diagnóstico em mãos, a criança deve receber o tratamento logo. Os pais precisam sair do hospital sabendo se o teste foi feito ou se eles devem procurar uma Unidade Básica de Saúde para realizá-lo (HADACHI S; 2017).
A coleta do exame é bem simples: basta uma gotinha de sangue retirada do calcanhar do bebê (FIGURA 2). O resultado demora cerca de sete dias para ficar pronto e, quando o resultado não apresenta alterações, ele já vai para o site ou para o correio. Quando surge algo diferente, nem sempre significa o positivo de uma doença. Nesse caso, a gente pede para que outra coleta seja realizada para ter a confirmação (HADACHI S; 2017).
FIGURA 2: Teste do Pezinho
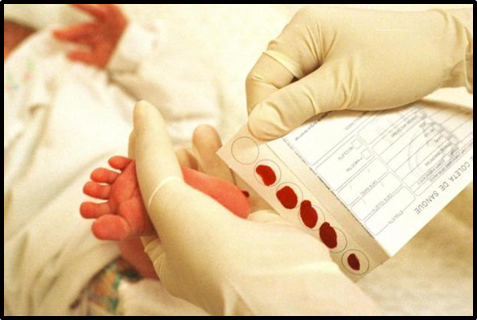
Fonte: NEWSLAB, 2022.
As crianças que nasceram antes das 37 semanas de gestação podem ser submetidas ao teste do pezinho, mas normalmente elas precisam fazer mais de uma coleta. O bebê prematuro deve repetir o exame com 120 dias de vida e, caso tenha recebido transfusão de sangue, 120 após a data da última transfusão (HADACHI S; 2017).
Obrigatório e gratuito em todo o território nacional desde 1992, o exame básico atualmente identifica seis enfermidades:
- Fenilcetonúria
- Hipotireodismo congênito
- Deficiência de biotinidase
- Fibrose cística
- Anemia falciforme
- Hiperplasia adrenal congênita
4.2.2 Hemoglobina Fetal
A hemoglobina Fetal, ou Hb F, tem importância essencial na fisiologia da distribuição de oxigênio durante a fase fetal que abrange o período entre o terceiro mês de gestação até o dia do nascimento. Nos dois primeiros meses de gestação, os eritrócitos dos embriões humanos têm três tipos de hemoglobinas conhecidas coletivamente por hemoglobinas embrionárias e identificadas pelas diferenças moleculares de suas globinas em Hb Gower-1 (ζ2/ε2), Hb Gower-2 (α2/ε2) e Hb Portland (ζ2/γ2), (NAOUM PC; 2005).
Essas hemoglobinas são sintetizadas por genes específicos em diferentes fases da evolução embrionária e com especificidades próprias para efetuarem rápidas trocas de oxigênio com tecidos que se modificam constantemente (NAOUM PC; 2005).
Assim, quando a estrutura anatômica e fisiológica do feto se torna definida a partir do terceiro mês de gestação, as hemoglobinas embrionárias são substituídas integralmente pela Hb F. A principal razão fisiológica para essa mudança de tipos de hemoglobinas se deve a duas causas, a primeira, devido ao fato do expressivo crescimento da massa corporal que necessita de constância na suplementação de oxigênio, e a segunda, por causa da restrição de oxigênio ofertado pela circulação materno-fetal (NAOUM PC; 2005).
Para superar o baixo teor de oxigênio que chega para o feto, o processo evolutivo dos mamíferos, notadamente o da espécie humana, desenvolveu a Hb F que tem como principal característica fisiológica a alta afinidade pelo oxigênio, que faz com que a liberação de oxigênio dos eritrócitos para os tecidos fetais seja lenta. A estrutura molecular da Hb F é composta por duas globinas alfa e duas gama (α2γ2) e sua concentração atinge entre 90 e 95% até o nascimento (NAOUM PC; 2005).
Entretanto, a partir do sexto mês do desenvolvimento do feto ocorre também, mas de forma muito discreta, as sínteses das hemoglobinas normais A (α2β2) e A2 (α2δ2), de tal forma que ao nascer as suas concentrações não ultrapassam 10% e 2%, respectivamente. O nascimento provoca no organismo do recém-nascido uma brusca interrupção genética para a síntese de Hb F e um rápido estímulo para a produção de Hb A, e como o processo da eritropoiese medular demora por volta de oito dias, essas variações somente serão perceptíveis a partir da primeira semana de vida do bebê. Mas de forma geral, o padrão normal para as hemoglobinas de um recém-nascido na primeira semana é o seguinte: Hb F (90 a 95%), Hb A (5 a 10%) e Hb A2 (0,5 a 2%) (FIGURA 3), (NAOUM PC; 2005).
FIGURA 3: Parâmetro de Hemoglobina Fetal
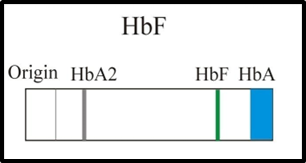
Fonte: LABPEDIA, 2022.
Num bebê com 30 dias de vida, por exemplo, é possível visualizar que a concentração da Hb F decresce e a da Hb A se eleva. Como a substituição das sínteses de globinas gama da Hb F por globinas beta da Hb A ocorre gradualmente, esse processo pode demorar entre dois e cinco meses. Dessa maneira, o padrão definitivo dos tipos e das concentrações de hemoglobinas para bebês é estabelecido a partir dos cinco meses de vida, conforme se segue: Hb A (96 a 98%), Hb A2 (2 a 4%) e Hb F (0 a 1%), (NAOUM PC; 2005).
A Hb F pode ser avaliada quantitativamente e qualitativamente. As análises quantitativas são as mais comuns e podem ser realizadas por meio de três testes diferentes: dosagens bioquímicas, eletroforeses e cromatografia por HPLC. Por outro lado, a análise qualitativa é realizada por um tipo de teste, a citologia intra-eritrocitária da Hb F, que determina se apenas alguns eritrócitos tem Hb F (distribuição heterogêna de Hb F) ou se todos eritrócitos as tem (distribuição homogênea da Hb F). Dosagens bioquímicas: há dois métodos, o de Singer(7) para concentrações acima de 10% de Hb F e o de Betke(2) para concentrações abaixo de 10%, ambos são conhecidos como teste de resistência alcalina para hemoglobina fetal (NAOUM PC; 2005).
Os dois métodos com excelentes graus de sensibilidade e reprodutibilidade técnicas estão fundamentados na capacidade química da Hb F ser resistente à desnaturação quando submetida à solução alcalina de NaOH 0,2N. As hemoglobinas A, S, C, entre outras, não são resistentes às soluções alcalinas e se desnaturam com facilidade. Pelo fato desses testes serem trabalhosos e com várias etapas de reações, os mesmos foram excluídos da rotina laboratorial e substituídos pelas eletroforeses ou cromatografia HPLC (NAOUM PC; 2005).
4.2.3 Hemoglobina S
A hemoglobina S é uma variante da hemoglobina encontrada nas pessoas originárias do Norte e Ocidente Africano, do Médio-Oriente, Índia e Bacia do Mediterrâneo. Isto se deve ao fato de que nestas áreas a malária ser endêmica e a Hb S conferir uma proteção relativa contra a doença (SÁ, J; 2018).
Chamamos de drepanocitose o fenômeno em que as hemácias deixam o seu formato bicôncavo e adquirem o formato de foice. A drepanocitose acontece na anemia falciforme (SÁ, J; 2018).
A variante Hb S está associada a uma mutação que ocorre no gene β-globínico, que codifica as cadeias ß da Hb. Um indivíduo pode ser heterozigótico para a mutação (AS), quando só um dos genes globínicos está mutado ou Homozigótico (SS), ou Heterozigótico composto (SC, SD, SO-Arab, S -talassemia) quando os dois genes beta globínicos estão afetados neste caso há doença – Drepanocitose. A pessoa com drepanocitose pode apresentar os seguintes sintomas (SÁ, J; 2018).
- Icterícia (cor amarela dos olhos);
- Dores intensas (braços, pernas, tórax e abdômen), lesões dos tecidos (baço, pulmões, fígado, rins) devido a má circulação do sangue, pois o formato irregular das hemácias atrapalha seu transporte nas veias;
- Síndrome mão-pé: nas crianças pequenas as crises de dor podem ocorrer nos pequenos vasos sangüíneos das mãos e dos pés, causando inchaço, dor e vermelhidão no local;
- Úlcera (ferida) de Perna: ocorre mais freqüentemente próximo aos tornozelos, a partir da adolescência. As úlceras podem levar anos para a cicatrização completa, se não forem bem cuidadas no início do seu aparecimento. Para prevenir o aparecimento das úlceras, os pacientes devem usar meias grossas e sapatos;
- As pessoas com Drepanocitose desenvolvem anemia (palidez), (SÁ, J; 2018).
FIGURA 4: Icterícia
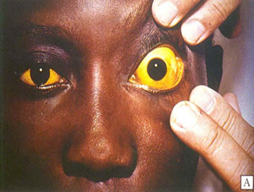
Fonte: NUPAD, 2022.
FIGURA 5: Síndrome mão-pé
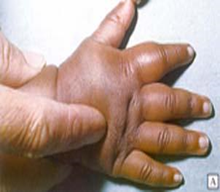
Fonte: MISODOR, 2022.
4.2.4 Eletroforese alcalina em acetato de celulose
A eletroforese alcalina é assim denominada devido ao fato do tampão utilizado para esse fim ter pH variável entre 8 e 9. Nessa faixa de pH a mutação que deu origem à Hb S (b 6 Glu ® Val) promoveu uma mudança de carga elétrica da molécula de Hb S, tornando-a menos negativa em relação à Hb A. Assim, quando amostras de sangue com diferentes genótipos são submetidas à eletroforese, a Hb S se move eletricamente de forma mais lenta que a Hb A (NAOUM PC et al; 2018).
Há dois tipos de eletroforese alcalina de hemoglobina que são usados com maior frequência em laboratórios: o acetato de celulose e a agarose. Os dois testes apresentam excelentes padrões de qualidade técnica, permitindo a qualificação dos principais genótipos de doenças falciformes, entretanto, a relação de hemoglobinas variantes que migram na mesma posição da Hb S, é necessário um meio de comprovação de que a hemoglobina variante em questão seja realmente a Hb S. O melhor meio para comprovar é a eletroforese ácida de hemoglobinas (NAOUM PC et al; 2018).
A eletroforese de hemoglobinas é de essencial importância no diagnóstico diferencial de anemias, microcitoses e hemólises, além de permitir análises familiares em parentes de portadores de hemoglobinas anormais. Seus resultados permitem o estabelecimento ou a exclusão de hemoglobinopatias e talassemias, constituindo importante e amplo procedimento diagnóstico, (NAOUM PC et al; 2018).
FIGURA 6: Cuba de Eletroforese

Fonte: PROLAB; 2022.
FIGURA 7: Resultado Eletroforese de Hemoglobina
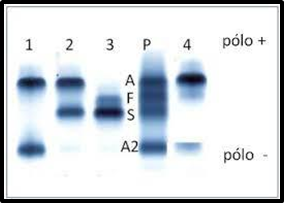
Fonte: MEDICINANET; 2022.
REFERÊNCIA:
- Hemoglobina A1 : > ou = 95,0%
- Hemoglobina Fetal :
- 1 a 7 dias : Até 84,0% 7 a 12 meses: Até 3,5%
- 8 a 60 dias : Até 77,0% 12 a 18 meses: Até 2,8%
- 2 a 4 meses: Até 40,0% Adulto: 0,0 a 2,0 %
- 4 a 6 meses: Até 7,0%
- Hemoglobina A2 : 1,5 a 3,5 % (NAOUM PC et al; 2018).
4.3 IMPORTÂNCIA DO TRATAMENTO PRÉVIO DA FALCIFORME EM CRIANÇAS
As crianças com DF apresentam risco de contrair infecções 400 vezes maior em relação à população infantil em geral. Por isso, é indicado rigoroso programa de prevenção, que alie o Calendário Nacional de Vacinação, estabelecido pelo MS, ao programa especial de vacinação para Haemophilusinfluenzae; hepatite B (recombinante); e Streptococcuspneumoniae (polissacáride e heptavalente) associado à profilaxia com penicilina injetável ou oral (ANVISA; 2002).
Essas vacinas já se encontram integradas ao Programa Nacional de Vacinação, e são utilizadas por todas as crianças brasileiras. Historicamente, os hemocentros têm sido a referência para o tratamento das doenças hematológicas, o que inclui as pessoas diagnosticadas com DF, mas em quatro estados (Acre, Mato Grosso do Sul, Rio Grande do Sul e Goiás) o centro de referência localiza-se em ambulatórios de especialidades ou nos hospitais universitários (ANVISA; 2002).
É importante lembrar que os neonatos diagnosticados como possíveis portadores de Doenças Falciformes deverão ser reavaliados laboratorialmente após o sexto mês de vida, e o estudo familiar dos possíveis casos deverão ser realizados.
4.4 TRATAMENTO E CURA DA ANEMIA FALCIFORME
O tratamento tem como objetivo a prevenção de crises e os problemas relacionados a elas. Pacientes com a doença falciforme fazem uso de ofolato ou ácido fólico para o controle da anemia e para auxiliar o organismo na produção de novos glóbulos vermelhos. O medicamento hidroxicarbamida também conhecido como Hidroxiureia também é utilizado no tratamento da doença falciforme, ele tem a função de aumentar a hemoglobina fetal, a mesma tem a capacidade de melhorar o transporte de oxigênio pelas hemácias, diminuindo assim a ocorrência de crises. (ANVISA; 2002).
O transplante de medula óssea pode curar anemia falciforme, a nova técnica substitui células doentes, evitando transfusões e a sobrecarga de ferro no sangue, em estudo realizado pela sociedade brasileira pode-se concluir que o transplante é o tratamento mais eficaz no combate a anemia falciforme, segundo os médicos que realizaram o estudo, com a prática de transplante as células doentes são substituídas e o paciente não precisa ser submetido a transfusões crônicas, e não recebe sobrecarga de ferro no sangue, consequência destas transfusões. Caracterizada por mudanças no formato das hemácias, cuja forma redonda passa a ser rígida e a ter aspecto de foice, o processo causa dois problemas, quando o organismo detecta a anormalidade, passa a destruir as hemácias, o que leva a anemia crônica. Já o formato da foice dificulta a circulação e facilita a formação de coágulos, aumentando o risco de AVC e de trombose (OLIVEIRA, 2014).
5. CONCLUSÃO
A partir do estudo realizado foi possível alcançar os objetivos propostos, aumentando o conhecimento sobre a anemia falciforme, suas características e diagnósticos. Trata-se de uma doença genética que, quando diagnosticada precocemente em recém nascidos ou crianças tem maior chance de cura através do transplante de medula óssea. O diagnóstico pode ser feito através do teste do pezinho na primeira semana de vida, pode ser feito outro teste de confirmação, como a eletroforese de hemoglobina. Por isso é de extrema importância que logo após o nascimento, seja feito o exame do teste do pezinho, para um diagnóstico imediato, assim que a doença for diagnosticada, iniciar o tratamento do portador da doença falciforme, para que os sintomas possam sem reduzidos e sua qualidade de vida melhorada.
REFERÊNCIAS BIBLIOGRÁFICAS
ANVISA (2002). In: Manual de Diagnóstico e Tratamento de Doenças Falciformes. Brasília,. 142p.
CEHMOB-MG (2014) Doença Falciforme em números – Setor de Gestão da Informação do Núcleo de Ações e Pesquisa em Apoio Diagnóstico da Faculdade de Medicina da UFMG (NUPAD), CEHMOB, Belo Horizonte, Disponível em: http://www.cehmob.org.br/?page_id=174#sthash.2trVbuN4.dpuf
HERRICK JB. (1910) Peculiar Elongated and Sickle-shaped Red Blood Corpuscles in a Case of Severe Anemia. Arch Int Med. v. 5, n. 517, p. 517-521.
GALIZA NETO GC, PITOMBEIRA MS. (2003) Aspectos moleculares da anemia falciforme. J Bras Patol Med Lab, v. 39, n.1, p. 51-56,.
LOBO C; MARRAVN; SILVA RMG. (2007) Crises dolorosas na doença falciforme. Rev Bras Hemato Hemoter, v.29, n.3, p. 247-258.
LOUREIRO, M.M; ROZENFELD, S. (2005) Epidemiologia de internações por doença falciforme no Brasil. Rev. Saúde Pública. Vol.39, n. 6, São Paulo.
NAOUM PC. (2000.) Interferentes Eritrocitários e Ambientais na Anemia Falciforme. Rev Bras Hematol Hemot. v. 22, n.1, p. 5-22.
NAOUM PC. (2012) Eletroforeses: Hemoglobinopatias, Proteínas Séricas, Lipoproteases, DNA – São Paulo; Santos.
SILVA YP; SILVA JF. (2006) A dor na criança com doença falciforme. In: Dor em Pediatria. Rio de Janeiro: Guanabara Koogan, cap. 15, p. 129-134.
SOUZA, J.C. (2009) Hematologia. 26ª Ed. Apostila publicada em abril de 2009.
HADACHI SÔNIA (2017) – Laboratório do Serviço de Triagem Neonatal da APAE – São Paulo
NAOUM PC. (2007) Rev. bras. hematol. Hemoter ;29(3):226-228
FRANCES T (2003) A Manual of Laboratory and Diagnostic Tests. Lippincott Williams & Wilkins.
NAOUM PC (2005) Stomatoyannopoulos G – Control of globin gene expression. During development and erythroid differentiation. Exp Hematol, 33: 259-271.
NAOUM PC et al (2018) Diagnóstico laboratorial das doenças das células falciforme. http://hemoglobinopatias.com.br/diagnostico-laboratorial-das-doencas-das-celulas-falciformes
SÁ, J (2018) O que é hemoglobina S? http://omundodapatologiaclinica.blogspot.com.br/2013/05/o-que-e-hemoglobina-s.html
ANVISA (2002) Manual de Diagnóstico e Tratamento de Doenças Falciformes, Brasília.
OLIVEIRA, R A. F. Antígenos Leucocitários Humanos (HLA) na Avaliação Imunológica para a Seleção de Receptor-Doador para Transplantes. 2014. 210 f.