REGISTRO DOI: 10.69849/revistaft/fa10202506091546
Najla Neme Dutra1
Zelio Fedatto Junior2
RESUMO
As doenças cardiovasculares representam um dos maiores desafios de saúde pública em todo o mundo, sendo responsáveis por uma parcela significativa da morbidade e mortalidade global. As estatinas são eficazes na redução de LDL colesterol, no entanto, uma grande parcela dos pacientes não reduz essa taxa satisfatoriamente ou não têm boa adesão ao tratamento devido aos efeitos colaterais. Além de as doenças cardiovasculares adquiridas, as hereditárias representam um grande problema de saúde, principalmente na população mais jovem. Diante desses problemas, novas estratégias para a elaboração de novos tratamentos – e até mesmo a cura – vem sendo amplamente explorado. Nesse contexto, abordagens de edição gênica, como CRISPR-Cas9, têm sido amplamente exploradas. A tecnologia CRISPR-Cas9 permite a edição precisa do genoma, oferecendo a possibilidade de corrigir mutações genéticas específicas associadas a doenças cardiovasculares hereditárias. Essa técnica tem sido utilizada em modelos animais e estudos pré-clínicos para corrigir mutações que levam a condições como a cardiomiopatia hipertrófica e outras doenças cardíacas genéticas. A terapia gênica mediada por CRISPR também está sendo explorada para tratar doenças cardiovasculares adquiridas, como a aterosclerose, oferecendo novas possibilidades para o tratamento da hipertensão. No entanto, apesar do potencial transformador da edição gênica, existem desafios significativos, como a entrega eficiente e segura dos componentes de edição gênica e a minimização de efeitos off-target. A integração de novas tecnologias pode oferecer soluções para esses desafios, abrindo novas fronteiras no tratamento das doenças cardiovasculares e melhorando a qualidade de vida dos pacientes. A presente revisão narrativa tem o objetivo explorar os avanços nas abordagens de edição gênica, como CRISPRCas9 no tratamento de doenças cardiovasculares.
Palavras-chave: Genética; Eventos Cardíacos; Edição de Genomas.
ABSTRACT
Cardiovascular diseases represent one of the greatest public health challenges worldwide, accounting for a significant portion of global morbidity and mortality. Statins are effective in reducing LDL cholesterol; however, a large portion of patients do not achieve satisfactory reduction or have poor adherence to treatment due to side effects. In addition to acquired cardiovascular diseases, hereditary conditions also pose a major health problem, especially among younger populations. In light of these issues, new strategies for developing treatments — and even potential cures — are being extensively explored. In this context, gene-editing approaches such as CRISPR-Cas9 have garnered significant attention. The CRISPR-Cas9 technology allows for precise genome editing, offering the possibility of correcting specific genetic mutations associated with hereditary cardiovascular diseases. This technique has been used in animal models and preclinical studies to correct mutations that lead to conditions such as hypertrophic cardiomyopathy and other genetic heart disorders. CRISPRmediated gene therapy is also being explored as a treatment for acquired cardiovascular diseases, such as atherosclerosis, presenting new possibilities for managing hypertension. However, despite the transformative potential of gene editing, significant challenges remain, such as the efficient and safe delivery of gene-editing components and the minimization of off-target effects. The integration of new technologies may offer solutions to these challenges, opening new frontiers in the treatment of cardiovascular diseases and improving patients’ quality of life. This narrative review aims to explore recent advances in geneediting approaches, such as CRISPR-Cas9, for the treatment of cardiovascular diseases.
Keywords: Genetics; Cardiac Events; Genome Editing.
Introdução
Doenças cardiovasculares (DCVs) incluindo doença cardíaca isquêmica e o acidente vascular cerebral (AVC) são a principal causa de morte globalmente sendo responsáveis por aproximadamente 20 milhões de mortes anualmente. Esse cenário representa um fardo significativo tanto para os sistemas de saúde quanto na qualidade de vida dos indivíduos afetados. Estima-se que aproximadamente três quartos das mortes associadas às DCVs ocorrem em países de baixa e média renda, o que evidencia a os impactos sociais dessas doenças. As implicações extrapolam o âmbito individual, afetando também famílias e economias nacionais, em virtude dos elevados custos com tratamento, reabilitação e perdas econômicas associadas à redução de produtividade (Di Ceasare et al., 2024).
A elevada prevalência das DCVs é impulsionada por uma interação multifatorial de elementos de risco clássicos, como hipertensão arterial sistêmica, dislipidemias, tabagismo, diabetes mellitus e obesidade (Mao et al., 2021). Embora avanços consideráveis tenham sido alcançados nas estratégias de prevenção e tratamento, fatores comportamentais, ambientais e metabólicos continuam a sustentar a crescente carga dessas enfermidades. Projeções baseadas nos dados do Global Burden of Disease (GBD) de 2019 demonstram que a prevalência global de DCVs pode praticamente dobrar até 2050, passando de 598 milhões de casos em 2025 para 1,14 bilhão, com um aumento estimado de 73,4% na mortalidade no mesmo período. Apesar da tendência de queda nas taxas padronizadas por idade de mortalidade e prevalência, reflexo de esforços em saúde pública e avanços terapêuticos, esses ganhos serão parcialmente neutralizados pelo envelhecimento populacional e pelo crescimento demográfico (Chong et al., 2024).
As estatinas têm sido a espinha dorsal da prevenção primária e secundária das DCVs há décadas. Seu mecanismo de ação baseia-se na inibição competitiva da enzima HMG-CoA redutase, etapa limitante da via mevalonato, responsável pela síntese hepática de colesterol. Essa inibição leva à redução da produção endógena de colesterol e ao consequente aumento de expressão dos receptores de LDL nos hepatócitos, promovendo maior depuração plasmática de LDL-colesterol (LDL-C). A magnitude da redução de eventos cardiovasculares é proporcional ao grau de redução de LDL-C, com estudos demonstrando uma diminuição dose-dependente entre 25% e 60% com o uso de estatinas como a atorvastatina9 (Nawrocki et al., 1995).
No entanto, apesar de sua eficácia consolidada, as terapias atualmente disponíveis apresentam limitações significativas (Abdul-Rahman et al., 2022). Uma meta-análise realizada por Boekholdt et al. mostrou que, entre os pacientes em uso de terapia com estatinas em altas doses, mais de 40% não atingiram a meta de LDL-C < 70 mg/dL. (Boekholdt et al., 2014), manifestada por efeitos adversos como miopatia e mioalgia, que podem acometer até 15% dos usuários (Newman et al., 2019). Além do mais, a adesão terapêutica a longo prazo frequentemente se mostra baixa, prejudicando os desfechos clínicos e a eficácia das estratégias de prevenção (Boekholdt et al., 2014).
Diante das limitações das terapias farmacológicas convencionais, como variabilidade na resposta terapêutica, efeitos adversos associados às estatinas e baixa adesão ao tratamento de longo prazo, as estratégias baseadas em manipulação genética têm ganhado destaque como abordagens promissoras no manejo das DCVs. Abordagens de edição gênica como CRISPR-Cas9, têm possibilitado a identificação de variantes genéticas associadas à susceptibilidade, gravidade e à resposta ao tratamento das DCVs (Bonowicz et al., 2025).
Além das DCVs adquiridas, é essencial considerar o impacto das doenças cardiovasculares de origem genética, que representam uma proporção relevante, especialmente em populações jovens (Bao et al., 1995). Dentre essas, destacam-se as cardiomiopatias hereditárias, como a cardiomiopatia hipertrófica (HCM), a cardiomiopatia dilatada (DCM) e a cardiomiopatia arritmogênica (ACM), geralmente herdadas por padrão autossômico dominante. Essas condições são causadas por mutações em genes que codificam proteínas estruturais do sarcômero, como MYBPC3, MYH7 e TTN. Tais mutações levam a alterações na contratilidade, estrutura e função cardíaca, podendo culminar em insuficiência cardíaca ou morte súbita. Nesses casos, a edição gênica surge como uma ferramenta promissora, com estudos experimentais demonstrando que a correção de mutações por edição de bases ou inativação seletiva de alelos mutantes pode restaurar a função cardíaca e prevenir o desenvolvimento da doença (Wilcox et al., 2018).
Portanto, a incorporação da medicina genômica ao manejo das DCVs hereditárias representa não apenas um avanço científico, mas uma nova fronteira no tratamento personalizado e preventivo de doenças cardiovasculares de alta gravidade. Sendo assim, essa revisão tem como objetivo explorar os avanços recentes em terapias genéticas e moleculares para o tratamento das doenças cardiovasculares.
Metodologia
Busca na base de dados
A busca bibliográfica foi realizada nas principais bases de dados científicas, incluindo PubMed, Scopus e Web of Science. Os descritores utilizados incluíram termos como “CRISPR/Cas9” OR “gene editing” OR “gene therapy” OR ‘genetic therapy”, “cardiovascular diseases” OR “heart diseases” OR “cardiovascular diseases”. A Figura 1 é a que melhor representa este esquema.
Critérios de seleção do estudo
Foram incluídos estudos que apresentassem aplicações experimentais da tecnologia CRISPR/Cas9 para investigar variantes genéticas relacionadas a doenças cardiovasculares. Foi dada a preferência a pesquisas que utilizassem modelos animais transgênicos ou células humanas derivadas de células-tronco pluripotentes induzidas (hiPSCs) para modelagem funcional. Artigos de revisão, estudos clínicos e pesquisas com métodos robustos de validação genética e fenotípica foram priorizados.
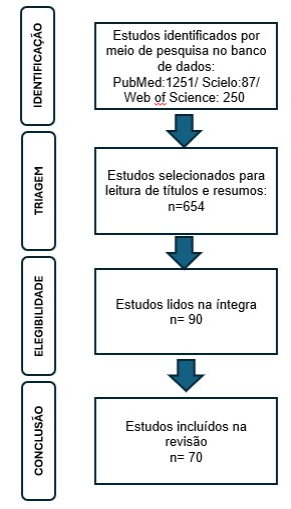
Figura 1. Fluxograma de inclusão de trabalhos no estudo
A terapia gênica é uma estratégia terapêutica que busca tratar ou prevenir doenças por meio da modificação da expressão genética nas células do paciente, e pode ocorrer por diferentes mecanismos: adição, substituição ou silenciamento gênico (Bak et al., 2018). Na adição gênica, um gene funcional é introduzido para compensar a ausência ou deficiência de um gene nativo, como ocorre em algumas doenças, como a imunodeficiência combinada severa (SCID). Há ainda, a substituição gênica, que por sua vez, visa corrigir uma mutação específica ao inserir uma cópia funcional do gene diretamente no local afetado do genoma, o que demanda ferramentas mais precisas, como as tecnologias de edição genômica. O silenciamento gênico tem como objetivo inativar genes cuja expressão exacerbada esteja associada a processos patológicos, podendo ser realizado com RNA interferente (RNAi), oligonucleotídeos antisense (ASOs) ou sistemas de edição como o CRISPR. Um exemplo clínico relevante dessa estratégia é o silenciamento do gene PCSK9 para redução dos níveis de colesterol LDL em pacientes com hipercolesterolemia familiar ou risco cardiovascular elevado (Li et al., 2023).
A edição genômica, embora relacionada à terapia gênica, representa uma abordagem mais precisa e dirigida, permitindo alterações pontuais no DNA, como inserções, deleções ou correções de bases específicas. As ferramentas mais utilizadas atualmente incluem o sistema CRISPR-Cas9, que atua como uma “tesoura molecular” guiada por RNA para cortar regiões-alvo do genoma, além de suas variantes, como Cas12, Cas13 e os editores de base ou de priming, que ampliam as possibilidades de intervenção com maior segurança e menor risco de erros nas bases alvo (Doudna et al., 2014).
O sistema de edição gênica CRISPR-Cas9 funciona por meio da entrega precisa de dois componentes essenciais: a proteína Cas9, responsável por cortar o DNA, e o RNA guia (gRNA), que direciona a Cas9 até a sequência-alvo no genoma. Essa entrega pode ser realizada por vetores virais, como adenovírus, que garantem uma boa penetração celular e expressão sustentada do sistema, ou por métodos físicos diretos, como eletroporação e microinjeção, que inserem os componentes diretamente no citoplasma ou núcleo celular (Mali et al., 2013). O gRNA é projetado para reconhecer uma sequência específica de DNA, formando um complexo com a Cas9 e guiando-a até o local exato onde a dupla hélice será rompida. A partir desse corte, inicia-se o processo de modificação genética. Essa interação entre Cas9 e gRNA, inspirada no sistema imune bacteriano natural, garante elevada especificidade e eficiência, tornando o CRISPR-Cas9 uma ferramenta versátil e poderosa na manipulação do genoma (Oude Blanke et al., 2016).
A terapia com AAV (vírus adeno-associado) consiste no uso de vetores virais modificados para entregar material genético terapêutico às células do paciente. Ela é amplamente utilizada em DCVs devido à sua capacidade de atingir com alta eficiência o tecido cardíaco, especialmente com o sorotipo AAV9. Essa abordagem permite a introdução de genes corretivos, silenciamento de genes patológicos ou entrega de ferramentas de edição genética, como o CRISPR, diretamente ao coração, oferecendo uma alternativa promissora para o tratamento de cardiomiopatias hereditárias e outras condições cardíacas genéticas (Zhang et al., 2022).
Diferentemente da terapia gênica tradicional, que geralmente adiciona genes de forma exógena, a edição genômica permite modificar diretamente a sequência original do DNA, promovendo intervenções mais duradouras e precisas (Bak et al., 2017). Ambas as abordagens vêm ganhando destaque no contexto da medicina de precisão e oferecem perspectivas promissoras para o tratamento de doenças cardiovasculares hereditárias, como as cardiomiopatias genéticas, e condições adquiridas, que estão associadas a fatores de risco modificáveis, como a dislipidemia.
Aplicação do CRISPR-Cas9 nas doenças cardiovasculares
Dislipidemias e aterosclerose
A aterosclerose é uma das principais causas de morbidade e mortalidade por DCVs no mundo, sendo responsável por cerca de 17,9 milhões de mortes anuais, segundo a OMS (WHO, 2021). Apesar da eficácia clínica de terapias para redução do colesterol LDL, como estatinas e inibidores de PCSK9, desafios como baixa adesão dos pacientes, alto custo e resposta insatisfatória em alguns casos limitam seu uso (Tonelli et al., 2011). Nesse contexto, o CRISPR-Cas9 tem se destacado como uma abordagem inovadora e promissora, permitindo modificações precisas no DNA para efeitos terapêuticos de longa duração com uma única intervenção. Em relação à aterosclerose, o alvo principal tem sido o gene PCSK9 que regula a degradação dos receptores de LDL (Cohen et al., 2006).
Estudos mostram que a inativação desse gene pode reduzir significativamente os níveis de LDL-C e o risco de eventos cardiovasculares. Com base nesses achados, inibidores de PCSK9 como Evolocumabe (Sabatine et al., 2017). e Alirocumabe (Schwartz, 2018) já estão aprovados para uso clínico, reforçando o potencial dessa via terapêutica.
Além dessas abordagens, novas fronteiras estão sendo exploradas com a transferência e edição genética, especialmente utilizando CRISPR-Cas9. A edição genética permite modificar permanentemente o gene PCSK9 no fígado, com o objetivo de desativá-lo e reduzir de forma duradoura a produção de LDLC. O exemplo mais notável é o VERVE-101, uma terapia de edição de base desenvolvida para silenciar o gene PCSK9 em hepatócitos por meio de uma única infusão intravenosa contendo um editor de adenina (base editing) encapsulado em nanopartículas lipídicas (VerveTherapeutics, 2024). Em e saios clínicos iniciais (Heart-1), pacientes com hipercolesterolemia familiar (HF) heterozigótica receberam doses ascendentes da terapia. Resultados preliminares mostraram reduções sustentadas de até 55% no LDL-C e reduções de até 84% nos níveis circulantes de PCSK9, com duração de até seis meses (Hooper et al., 2024).
No entanto, efeitos adversos também foram observados, incluindo reações infusionais leves a moderadas, elevação transitória de enzimas hepáticas (ALT) e eventos cardiovasculares isolados, como infarto do miocárdio e arritmias. Esses efeitos podem estar relacionados ao impacto da inibição de PCSK9 sobre proteínas como CD36 e VLDL-R, que estão envolvidas no metabolismo de ácidos graxos e podem afetar tecidos sensíveis como músculo cardíaco e plaquetas (Puccini et al., 2022).
A terapia gênica mediada por vetores virais, utilizando os AAVs se mostram como uma abordagem promissora para o tratamento da HF (Kassim et al., 2010), um fator de risco importante para a aterosclerose e outras doenças cardiovasculares. Dentre os vetores virais utilizados, os vírus AAVs se destacam por sua alta eficiência de transdução, baixa imunogenicidade e capacidade de promover expressão sustentada do gene terapêutico (Tirronen et al., 2019). Essa tecnologia tem sido explorada em diferentes modelos pré-clínicos como uma estratégia de intervenção duradoura para o controle dos níveis lipídicos.
Em um estudo experimental conduzido por Peter Tontonoz e colaboradores (2017), foi utilizado um vetor AAV8 contendo a sequência de RNA não codificante LeXis (liver-expressed liver X receptor-induced sequence), sob controle do promotor hepatoespecífico TBG (thyroxine-binding globulin), com o objetivo de avaliar seus efeitos terapêuticos em um modelo murino de HF. A expressão sustentada de LeXis induzida pelo vetor resultou na supressão do gene regulador lipídico Srebp2, levando à redução da síntese de lipídios hepáticos. Análises histológicas confirmaram uma diminuição expressiva de lipídios no fígado dos animais tratados, e exames bioquímicos demonstraram reduções significativas nos níveis séricos de colesterol total e triglicerídeos.
O Vupanorsen (também conhecido como AKCEA-ANGPTL3-L Rx) representa uma abordagem inovadora na medicina de precisão voltada ao tratamento de dislipidemias complexas e risco cardiovascular residual. Trata-se de um ASOs conjugado com N-acetilgalactosamina (GalNAc), o que direciona sua ação especificamente para o fígado – o órgão central no metabolismo lipídico (Gaudet et al., 2020). Sua principal função é inibir a síntese da proteína angiopoietina-like 3 (ANGPTL3), um regulador fundamental das lipoproteínas plasmáticas. Pesquisas clínicas demonstraram que a inativação de ANGPTL3 – seja por meio de anticorpos monoclonais ou por ASOs como o Vupanorsen – leva a reduções significativas nos níveis plasmáticos de LDL-C e triglicerídeos, especialmente em pacientes com dislipidemias de origem genética ou complexas (Kersten, 2021).
Essa abordagem é interessante, principalmente para pessoas que, mesmo sob tratamento com estatinas e outros agentes hipolipemiantes, mantêm risco cardiovascular elevado devido à presença de lipídios aterogênicos residuais. Particularmente interessante é a aplicação do Vupanorsen em pacientes com lipodistrofia parcial familiar (FPLD), uma condição metabólica rara caracterizada por perda seletiva de tecido adiposo subcutâneo, resistência à insulina e hipertrigliceridemia grave (Foss-Freitas et al., 2021). Ao reduzir ANGPTL3, o Vupanorsen pode melhorar o perfil lipídico e metabólico desses pacientes, oferecendo alívio para um conjunto complexo de distúrbios metabólicos que, até então, contavam com poucas opções terapêuticas eficazes. Além disso, níveis naturalmente baixos de ANGPTL3 foram associados a perfis lipídicos mais favoráveis – com menores concentrações de triglicerídeos, LDL e outras partículas ricas em apolipoproteína B – o que reforça o potencial da inibição dessa proteína como estratégia terapêutica (Ando et al., 2003).
Miocardiopatias genéticas
As cardiomiopatias hereditárias, como a dilatada (DCM), hipertrófica (HCM) e arritmogênica (ACM), são comumente herdadas de forma autossômica dominante. No entanto também existem casos com herança autossômica recessiva, ligada ao X ou mitocondrial. Por exemplo, a HCM é causada por mutações em genes sarcoméricos que geralmente atuam por efeito dominantenegativo ou haploinsuficiência, levando a uma ativação excessiva dos miofilamentos, hiper contratilidade do músculo cardíaco e aumento no consumo de energia (Cahill et al., 2013).
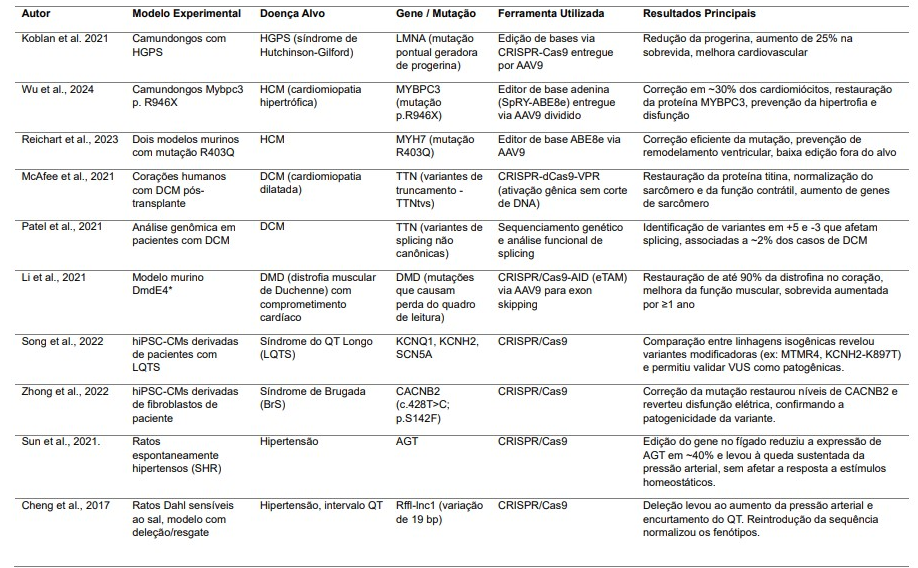
Koblan e colegas (2021) demonstraram que a edição de bases in vivo foi eficaz para tratar a síndrome de Hutchinson-Gilford (HGPS) em camundongos. HGPS é uma doença genética rara caracterizada por envelhecimento acelerado, com expectativa de vida média de 14 anos, sendo as complicações cardiovasculares a principal causa de morte. A maioria dos casos é causada por uma mutação pontual dominante no gene LMNA, que gera uma proteína truncada (incompleta) chamada progerina. Essa proteína altera a estrutura e função do núcleo celular, levando à senescência precoce e morte celular.
Estudos anteriores usaram edição gênica para introduzir mutações do tipo indel (do inglês insertion e deletion), reduzindo a expressão de lamin A/progerina sem afetar lamin C. A entrega do CRISPR-Cas9 via AAV9 em camundongos neonatais com HGPS levou à redução parcial da progerina e aumento de 25% na sobrevida (Tabela 1).
Dentre as miocardiopatias genéticas, há a HCM, uma doença que é herdada de forma autossômica dominante e, em cerca de 70% dos casos com mutações em genes sarcoméricos, envolve os genes MYBPC3 e MYH7. O MYBPC3, além de estar associado à HCM, também está implicado em outras cardiomiopatias, como a restritiva, dilatada e a não compactação do ventrículo esquerdo. Esse gene codifica a proteína cMyBP-C, um componente estrutural essencial do músculo cardíaco, que interage com actina, miosina e titina, contribuindo para a estabilidade e função do sarcômero (Toepfer et al., 2019).
Em um estudo de Wu e colegas (2024) foi criado um modelo murino com mutação Mybpc3 (p.R946X) para simular HCM grave. Utilizaram um editor de base adenina (SpRY-ABE8e), entregue via AAV9 dividido, para corrigir a mutação in vivo. A edição corrigiu cerca de 30% dos cardiomiócitos e restaurou até 110% da proteína MYBPC3. O tratamento preveniu hipertrofia, disfunção cardíaca, fibrose e normalizou a expressão gênica. Efeitos colaterais foram mínimos, com baixa edição fora do alvo (Tabela 1).
Esse estudo corrobora com resultados anteriores, no qual pesquisadores desenvolveram um editor de base dividido em dois vetores AAV9 para corrigir a mutação R403Q associada à HCM (Tabela 1). Em dois modelos murinos, a edição com ABE8e corrigiu eficientemente a mutação nas células cardíacas, prevenindo hipertrofia, remodelamento ventricular, fibrose e alterações no transcriptoma. A edição foi específica para cardiomiócitos, com baixa edição fora do alvo. Uma segunda dose aumentou a correção em átrios, mas não em ventrículos. Alternativamente, a inativação da cópia mutante via Cas9 também impediu o desenvolvimento da HCM (Reichart et al., 2023).
As variantes de truncamento no gene TTN (TTNtvs) são a alteração genética mais comum identificada em indivíduos com DCM, uma doença associada à altas taxas de morbidade e mortalidade. Essas variantes reduzem os níveis da proteína titina normal, levam à produção de proteínas truncadas e comprometem a estrutura e a função dos sarcômeros, unidades responsáveis pela contração muscular no coração (Akinrinade et al., 2019). Em um estudo de McAfee et al. (2021), foi avaliado a presença de variantes TTNtvs no coração de 184 pacientes com DCM que haviam passado por transplante. O estudo confirma que proteínas truncadas da titina estão presentes em corações humanos com TTNv, que há menos titina funcional nesses casos, e que a doença resulta tanto da perda de função (haploinsuficiência) quanto da ação tóxica das proteínas truncadas (efeito dominante-negativo).
A técnica de CRISPR-dCas9-VPR, diferentemente da tradicional, não corta o DNA, mas ativa a expressão de genes. Nesse contexto, foi observado que a ativação do gene TTN por CRISPR-dCas9 direcionada ao promotor ou a regiões regulatórias próximas por interações tridimensionais do DNA, restaurou os níveis da proteína titina em células com TTNtvs. O aumento da proteína titina normalizou a estrutura do sarcômero e a função contrátil, mesmo com o aumento das proteínas truncadas. Além disso, houve aumento na expressão de genes relacionados à montagem de miofibrilas e sarcômeros (Tabela 1). Ou seja, a ativação de TTN por CRISPR corrigiu os déficits funcionais causados por TTNtvs, apoiando a ideia de haploinsuficiência como o principal mecanismo da doença em variantes heterozigotas. Essa abordagem mostra potencial terapêutico para tratar grande parte dos casos de DCM relacionados a TTNtvs (McAfee et al., 2021).
Além das mutações que causam perda de função diretamente por meio de variantes de truncamento, o gene TTN também é suscetível a alterações no processamento de RNA, especialmente através de mutações que afetam o splicing. O splicing alternativo é um mecanismo essencial que permite a geração de diferentes isoformas proteicas a partir de um único gene (Tabela 1). Esse processo é regulado por elementos cis (enhancers e silencers) que ativam ou inibem o corte e junção dos exons durante a maturação do RNA mensageiro, em conjunto com proteínas reguladoras trans, como a família de proteínas SR ou as hnRNPs. Erros nesse mecanismo, especialmente no coração, podem levar à produção de proteínas aberrantes ou não funcionais, contribuindo para doenças cardiovasculares hereditárias. No caso da DCM, TTNtvs são responsáveis por até 20% dos casos idiopáticos (Patel et al., 2021).
No entanto, estudos recentes ampliaram a compreensão do impacto genético além das mutações clássicas de perda de função. Um estudo demonstrou que variantes raras localizadas em regiões de splicing próximas a exons altamente expressos do TTN, mas que não envolvem os dinucleotídeos invariantes dos sítios canônicos de splice donor e aceptor, também podem alterar o processamento do RNA. Essas variantes, chamadas de variantes de splicing não canônicas, foram encontradas em cerca de 2% dos pacientes com DCM, sugerindo um impacto patogênico relevante. Especificamente, alterações nos nucleotídeos +5 do sítio doador e -3 do sítio aceptor foram significativamente associadas a efeitos no splicing (P = 4,4×10⁷ e P = 0,002, respectivamente) (Patel et al., 2021).
Assim como as mutações no gene TTN podem levar a doenças cardiovasculares hereditárias devido a erros no processamento de RNA e splicing alternativo, mutações no gene DMD também resultam em distúrbios musculares graves, como a distrofia muscular de Duchenne (DMD), devido à perda de função da proteína distrofina. A DMD é causada pela ausência de distrofina, levando à degeneração progressiva dos músculos esqueléticos e cardíacos, sendo a insuficiência cardíaca a principal causa de morte. O gene DMD é composto por 79 éxons e codifica a distrofina, proteína essencial para a integridade das fibras musculares. Mutações que causam a distrofia muscular de DMD frequentemente resultam na perda do quadro de leitura do RNA mensageiro, levando à ausência da distrofina funcional. Em contrapartida, mutações que preservam o quadro de leitura causam a distrofia muscular de Becker (BMD), uma forma mais branda da doença, pois permitem a produção de uma distrofina encurtada, mas parcialmente funcional (Koenig et al., 1989).
Essas mutações no gene DMD inspiraram o desenvolvimento da estratégia de exon skipping, técnica que “pula” éxons mutados e permite a produção de uma forma funcional da proteína. Essa técnica visa restaurar o quadro de leitura do RNA ao promover a exclusão programada de certos éxons durante o processamento do pré-mRNA. Essa técnica utiliza ASOs e se ligam a regiões específicas do RNA e mascaram sinais de splicing, induzindo o “pulo” do éxon-alvo. A exclusão do éxon 51, por exemplo, é a abordagem mais estudada e pode beneficiar cerca de 13% dos pacientes com DMD. Há ainda interesse em estratégias de exclusão múltipla de éxons, especialmente entre os éxons 45 a 55 – região onde se concentram muitas mutações e cuja remoção simultânea poderia tratar até 63% dos casos (Aartsmarus et al., 2009).
No entanto, essa abordagem enfrenta desafios técnicos, como a baixa eficiência e a complexidade do splicing em genes longos como o DMD, cuja transcrição completa pode durar até 16 horas (Béroud et al, 2007). Estudos recentes com ASOs modificados, como dendrímeros octaguanidínicos, têm mostrado resultados promissores em aumentar a eficácia do multiple exon skipping, o que amplia o potencial terapêutico dessa técnica. Em um novo modelo murino (DmdE4*), que reproduz características humanas da DMD, pesquisadores testaram a edição de bases citidínica para promover exon skipping. No contexto da experimentação animal, usando AAV9 para entregar o CRISPR/Cas9-AID (eTAM), foi possível restaurar até 90% da distrofina no coração, prevenir o remodelamento ventricular, melhorar a função muscular e aumentar a sobrevida dos camundongos, com efeitos duradouros por pelo menos um ano (Tabela 1). Esses achados destacam o potencial do exon skipping mediado por CRISPR como terapia promissora para tratar a cardiomiopatia associada à DMD (Li et al., 2021).
Arritmias genéticas
As arritmias genéticas são distúrbios herdados que afetam o sistema elétrico do coração, aumentando o risco de eventos graves como síncope e morte súbita, mesmo em indivíduos com coração estruturalmente normal. Entre as mais conhecidas estão a Síndrome do QT Longo (LQTS), caracterizada por uma repolarização ventricular prolongada que predispõe a arritmias ventriculares (Tester et al., 2014), e a Síndrome de Brugada (BrS), marcada por alterações específicas no eletrocardiograma e maior risco de fibrilação ventricular, especialmente em homens jovens (Brugada et al., 2018). Ambas são causadas por mutações em genes que codificam canais iônicos cardíacos e podem ser investigadas em modelos celulares derivados de células-tronco pluripotentes induzidas.
A LQTS é uma doença cardíaca hereditária causada principalmente por mutações em genes que codificam canais iônicos, como KCNQ1, KCNH2 e SCN5A, responsáveis pela repolarização do potencial de ação cardíaco. Essas mutações prolongam a duração do intervalo QT no eletrocardiograma, aumentando o risco de arritmias graves e morte súbita cardíaca (Goldenberg et al., 2008).
Cardiomiócitos podem ser originados de células-tronco pluripotentes induzidas de pacientes com diferentes variantes genéticas associadas a LQTS, como LQT1, LQT2 e LQT3. A edição genética via CRISPR/Cas9 permitiu a criação de linhagens isogênicas com e sem as mutações específicas, possibilitando comparar fenótipos celulares e estudar modificadores genéticos que influenciam a expressão clínica da síndrome (Tabela 1). Entre os principais achados, destacam-se a identificação de variantes modificadoras, como MTMR4 e KCNH2-K897T, que alteram a gravidade dos sintomas mesmo em indivíduos com a mesma mutação principal, além da validação de variantes de significado incerto (VUS) como patogênicas (Song et al., 2022).
A tecnologia CRISPR/Cas9 representa uma promissora abordagem para tratamento no futuro, no entanto, há muitos desafios a serem enfrentados. Em um estudo, no qual a edição gênica foi aplicada em ratos recém-nascidos O AAV9, conseguiu entregar a Cas9 em 60–70% dos cardiomiócitos, mas menos de 15% foram efetivamente editados geneticamente com uma aplicação. Isso se dá provavelmente se deve à menor atividade de remodelação da cromatina, deixando o DNA “inacessível” em células pós-mitóticas. Portanto, a ineficiência da edição genética ainda é uma das principais limitações do CRISPR/Cas9 (Johansen et al., 2017).
Ainda no contexto das arritmias genéticas, a BrS é uma canalopatia cardíaca hereditária que aumenta o risco de arritmias ventriculares fatais e morte súbita, especialmente em homens jovens. Os sintomas podem incluir síncope, convulsões e respiração agônica noturna, com ECG característico mostrando elevação do segmento ST nas derivações precordiais direitas. Sua prevalência é de cerca de 0,05% mundialmente, sendo mais comum em homens, com arritmias frequentemente ocorrendo durante o sono ou repouso (Vutthikraivit et al., 2018). Geneticamente, a BrS está associada a mutações em mais de 40 genes, principalmente no gene SCN5A (canal de sódio). No entanto, mutações em genes de canais de cálcio, como CACNB2, também têm sido implicadas, embora sua relação com o fenótipo ainda não seja bem compreendida. Compreender as bases genéticas da BrS, especialmente além do gene SCN5A, é essencial para o avanço de abordagens terapêuticas personalizadas.
Nesse contexto, estudos funcionais utilizando modelos celulares derivados de pacientes têm se mostrado uma abordagem importante para elucidar os efeitos de variantes raras, como demonstrado na pesquisa conduzida por Zhong e colaboradores. Estes autores investigaram um caso de BrS, que partir de fibroblastos da pele do paciente, os pesquisadores geraram hiPSCs, que foram subsequentemente diferenciadas em cardiomiócitos, permitindo a modelagem funcional da doença in vitro em um contexto celular personalizado. A mutação no CACNB2 foi inicialmente identificada por sequenciamento Sanger, após análise dos exons de diversos genes já relacionados à BrS. Para determinar se essa mutação era causativa, a equipe realizou a correção genética da variante utilizando CRISPR/Cas9, com a introdução de um complexo ribonucleoproteico (Cas9 + sgRNA) e um oligonucleotídeo doador de fita simples (ssODN) para direcionar a correção precisa do alelo mutante (Tabela 1). Clones isogênicos corrigidos foram obtidos e validados por sequenciamento, além de testados quanto à manutenção da pluripotência e estabilidade genômica (Zhong et al., 2022).
Nos cardiomiócitos derivados das hiPSCs do paciente, foi observada uma redução nos níveis da proteína CACNB2, compatível com um mecanismo de haploinsuficiência – condição em que uma única cópia funcional do gene não é suficiente para manter a função celular normal. Após a correção da mutação, os níveis de expressão de CACNB2 foram restaurados, e as anormalidades eletrofisiológicas previamente detectadas nas células do paciente foram revertidas. Esses achados forneceram evidência funcional direta de que a mutação S142F em CACNB2 era patogênica e responsável pela disfunção elétrica associada à BrS nesse caso. O estudo destaca não apenas a utilidade das hiPSCs para modelar doenças cardíacas hereditárias, mas também o potencial terapêutico da edição genética por CRISPR/Cas9 na correção de variantes causadoras de doenças, abrindo caminho para abordagens personalizadas no tratamento de canalopatias cardíacas (Zhong et al., 2022).
Hipertensão
A hipertensão afeta mais de 1 bilhão de pessoas no mundo. As complicações da hipertensão incluem AVC, insuficiência renal, hipertrofia cardíaca, infarto do miocárdio e insuficiência cardíaca (Dorans et al., 2017). Apesar do desenvolvimento de terapias anti-hipertensivas eficazes, o número de pessoas com hipertensão não controlada continua a aumentar, seja por falha no tratamento – geralmente estatinas – ou seja por falta de adesão à medicação. Esta última ocorre frequentemente devido ao incômodo de tomar vários medicamentos, várias vezes ao dia ou com efeitos colaterais frequentes. Como resultado, há grande interesse no desenvolvimento de novas terapias que possam proporcionar controle sustentado da hipertensão com menos efeitos adversos (Abdul-Rahman et al, 2022).
Nesse contexto, autores investigaram os efeitos terapêuticos do CRISPR/Cas9 sobre o sistema renina-angiotensina (SRA), um dos principais reguladores da pressão arterial e do equilíbrio hidrossalino. O alvo foi o gene do angiotensinogênio (AGT), precursor essencial na cascata de produção da angiotensina II, um potente vasoconstritor. Inicialmente, demonstraram a eficácia do CRISPR/Cas9 na edição do gene AGT in vitro, evidenciando sua capacidade de induzir modificações genéticas específicas e eficientes (Sun et al., 2021). Posteriormente, aplicaram a técnica em um modelo animal – ratos espontaneamente hipertensos (SHR) – amplamente utilizados na pesquisa sobre hipertensão (Tabela 1). A edição do gene AGT no fígado, principal local de sua expressão, resultou em uma redução de cerca de 40% na expressão do gene nos hepatócitos. Essa ablação parcial foi suficiente para induzir uma diminuição significativa e sustentada da pressão arterial nos animais. Importante destacar que, mesmo com a redução do AGT, os animais mantiveram respostas fisiológicas apropriadas a desafios como a depleção de sódio e a administração de furosemida, indicando preservação dos mecanismos homeostáticos. Esses achados sugerem que a edição parcial do gene AGT via CRISPR/Cas9 pode representar uma abordagem terapêutica eficaz e duradoura para o controle da hipertensão arterial, sem comprometer respostas adaptativas essenciais (Waghulde et al., 2018).
Pesquisadores utilizaram o CRISPR/Cas9 para manipular especificamente a região do RNA longo não codificante Rffl-lnc1, localizada dentro de um locus quantitativo associado à pressão arterial e ao intervalo QT em ratos Dahl sensíveis ao sal (Tabela 1). Foram gerados modelos de rato com diferentes deleções na região crítica de 19 pares de bases, observando-se que essas alterações causaram um encurtamento significativo do intervalo QT e aumento da pressão arterial, além de hipertrofia cardíaca, confirmando a relevância funcional do Rffl-lnc1 na regulação cardiovascular. Para validar a causalidade da variação de 19 bp, foi criado um modelo de “resgate” genético, no qual a sequência foi reinserida na linhagem congenicamente resistente (S.LEW), o que corrigiu os fenótipos alterados, normalizando pressão arterial e intervalo QT (Cheng et al., 2017).
A combinação de técnicas moleculares como a microinjeção de RNA guia e Cas9 em embriões, sequenciamento para validação das edições, e análises funcionais por eletrocardiograma e ecocardiografia permitiu demonstrar que essa pequena alteração no RNA não codificante é tanto necessária quanto suficiente para regular a função cardiovascular, destacando a importância dos elementos não codificantes no controle genético da hipertensão e dos riscos cardíacos associados (Cheng et al., 2017). Este estudo complementa as descobertas anteriores sobre os determinantes genéticos da hipertensão ao demonstrar, por meio de modelos de rato geneticamente editados com CRISPR/Cas9, que uma pequena variação de 19 pares de bases em um RNA longo não codificante (Rffl-lnc1) é suficiente para modular tanto a pressão arterial quanto o intervalo QT – dois importantes parâmetros cardiovasculares. Assim como na Síndrome de Brugada, em que uma mutação pontual no gene CACNB2 levou à disfunção elétrica cardíaca, este trabalho reforça o papel funcional direto de variantes genéticas específicas, mesmo em regiões não codificantes do genoma, como determinantes de doenças cardiovasculares (Cheng et al., 2017).
Juntos, os estudos de Sun et al. (2021), Waghulde et al. (2018) e Cheng et al. (2017) destacam como a combinação de modelagem funcional com células-tronco ou edição genética em animais pode esclarecer mecanismos moleculares complexos e abrir caminho para abordagens terapêuticas personalizadas no tratamento de canalopatias e hipertensão de origem genética.
Ensaios clínicos em andamento
A crescente compreensão dos mecanismos moleculares que envolvem a regulação cardiovascular, especialmente aqueles mediados por variantes genéticas e elementos não codificantes, tem impulsionado o desenvolvimento de terapias inovadoras, agora em fase de investigação clínica. Nesse contexto, ensaios clínicos em andamento têm explorado abordagens terapêuticas de última geração, como a inibição de PCSK9 por RNA interferente (inclisiran). Embora a aplicação direta dessas ferramentas no tratamento da hipertensão essencial ainda esteja em fase pré-clínica, os avanços em terapias gênicas demonstram o potencial translacional dessas estratégias e pavimentam o caminho para futuras intervenções personalizadas na prática clínica (Oostveen et al., 2023).
Inclisiran é uma terapia baseada em RNA de interferência (RNAi) desenvolvida com o objetivo de reduzir a expressão do gene PCSK9, cuja proteína desempenha papel crucial na regulação dos níveis plasmáticos de LDL-C. Após extensas modificações químicas e otimizações estruturais, foi desenvolvido o composto ALN-PCSsc, posteriormente denominado inclisiran, que utiliza a tecnologia de conjugação com ácido N-acetilgalactosamina (GalNAc) para facilitar a entrega seletiva ao fígado por via subcutânea, otimizando a eficácia terapêutica e a biodisponibilidade (Fitzgerald et al., 2014).
A avaliação inicial da segurança, tolerabilidade e eficácia farmacodinâmica de Inclisiran em seres humanos foi realizada em um ensaio clínico de fase I, publicado em 2017 (Fitzgerald et al., 2017). Nesse estudo, participaram voluntários saudáveis com níveis de LDL-C ≥ 100 mg/dL, os quais foram randomizados em uma proporção de 3:1 para receber Inclisiran ou placebo. O protocolo contemplou fases de dose única ascendente (variando entre 25 e 800 mg) e múltiplas doses (com esquemas de administração semanal, quinzenal ou mensal), com ou sem uso concomitante de estatinas. Os resultados demonstraram que doses únicas de 300 mg ou mais foram capazes de reduzir os níveis circulantes de PCSK9 em até 75%, com redução correspondente de aproximadamente 51% nos níveis de LDL-C. Destaca-se que tais reduções foram sustentadas por um período superior a seis meses após a administração da dose única, evidenciando o potencial de um regime terapêutico semestral. Além disso, o perfil de segurança foi considerado favorável, com a ocorrência apenas de eventos adversos leves a moderados, sem relatos de eventos graves ou descontinuações associadas ao tratamento (Fitzgerald et al., 2014).
A terapia VERVE-101 utiliza RNA mensageiro codificando uma adenina base editor (ABE) e um RNA guia específico para o gene da PCSK9. Essa combinação é encapsulada em nanopartículas lipídicas para administração sistêmica, realizada por infusão intravenosa única. O mecanismo de ação envolve a substituição de um par de bases adenina-timina (A/T) por guaninacitosina (G/C), levando à interrupção do sítio doador de splicing do gene PCSK9 no fígado e, consequentemente, à sua inativação. Estudos pré-clínicos em primatas e camundongos demonstraram que essa edição genômica é eficaz na redução prolongada dos níveis de PCSK9 e LDL-C, com efeitos duradouros superiores a um ano (Takahiro et al., 2024).
A fase clínica inicial da terapia VERVE-101 foi avaliada no ensaio HEART1 (NCT05398029), um estudo de fase 1b, aberto e de escalonamento de dose, que buscou investigar a segurança e tolerabilidade da terapia em humanos. Os primeiros resultados foram apresentados no congresso Scientific Sessions 2023 da American Heart Association. Nesse estudo, dez participantes com hipercolesterolemia familiar heterozigótica (HeFH), predominantemente do sexo masculino e com média de idade de 54 anos, receberam uma única infusão intravenosa de VERVE-101 em doses variando de 0,1 a 0,6 mg/kg. Esses indivíduos apresentavam níveis médios de LDL-C de 193 mg/dL e histórico cardiovascular importante, incluindo intervenções como revascularização miocárdica cirúrgica ou percutânea (Lee et al., 2023).
Os resultados preliminares foram animadores: reduções de LDL-C de 39% a 55% foram observadas nas doses consideradas terapêuticas (0,45 e 0,6 mg/kg), com reduções sustentadas por até 180 dias em um dos pacientes. Além disso, os níveis circulantes de PCSK9 também diminuíram significativamente. Os eventos adversos relatados foram, em geral, leves e transitórios, como reações à infusão e elevações reversíveis nas enzimas hepáticas (ALT), com níveis de bilirrubina dentro da normalidade. Três eventos adversos cardiovasculares graves ocorreram: um óbito por parada cardíaca considerado não relacionado ao tratamento, e dois eventos em um mesmo paciente – infarto do miocárdio logo após a infusão (potencialmente relacionado, devido à proximidade temporal) e taquicardia ventricular não sustentada semanas depois, considerado não relacionado. O comitê independente de monitoramento de dados e segurança concluiu que esses eventos estavam de acordo com o perfil clínico grave dos participantes, todos com doença cardiovascular estabelecida (Lee et al., 2023).
Apesar dos desafios inerentes à edição gênica in vivo – como possíveis efeitos fora do alvo, incertezas quanto às consequências de longo prazo e considerações éticas – os resultados dessa primeira aplicação clínica em humanos são promissores. A terapia gênica baseada em CRISPR representa uma abordagem inovadora e possivelmente definitiva para o tratamento de pacientes com hipercolesterolemia familiar e risco cardiovascular elevado. A continuidade dos ensaios clínicos será fundamental para confirmar a segurança, eficácia e aplicabilidade dessa tecnologia na prática clínica.
Desafios e considerações éticas
Dilemas regulatórios e de patentes
O uso do CRISPR-Cas9 levanta desafios regulatórios complexos. Uma das principais dificuldades é rastrear e controlar organismos geneticamente modificados (OGMs) fora do laboratório. Além disso, há disputas sobre a patente da tecnologia CRISPR, especialmente quanto ao uso terapêutico em humanos. A patente mais conhecida foi concedida à Caribou Biosciences em disputa com Feng Zhang. (Donohoue et al., 2018, Duardo-Sanchez-, 2017)
Edição genética para fins terapêuticos em células somáticas
O uso de CRISPR-Cas9 em células somáticas humanas (ou seja, não hereditárias) está crescendo rapidamente como forma de tratar doenças genéticas, como hipercolesterolemia familiar, anemia falciforme e certos tipos de câncer. Essas aplicações geralmente são mais aceitas do ponto de vista ético, pois os efeitos genéticos não são transmitidos às futuras gerações e são realizados com consentimento informado dos pacientes (De Graeff et al., 2019).
Edição genética na linhagem germinativa
A edição genética em células germinativas humanas (embriões, óvulos e espermatozoides), embora teoricamente possa eliminar doenças hereditárias, gera grandes preocupações éticas e científicas. Os principais pontos são: a) Riscos de mutações fora do alvo (off-target): essas alterações não intencionais podem causar defeitos graves ou até doenças fatais. Estudos mostram que os embriões são mais suscetíveis a essas mutações que células adultas (Roh et al., 2018); b) Transmissão hereditária de mutações: qualquer erro ou efeito adverso será transmitido para futuras gerações, tornando os riscos ainda mais significativos (Shinwary et al., 2017); c) Mosaicismo genético: pode ocorrer quando a edição genética é incompleta, resultando em células com diferentes genomas no mesmo organismo. Isso pode gerar doenças genéticas como Síndrome de Down ou Klinefelter (Otieno, 2015); d) Consentimento informado: não há como obter consentimento direto de embriões, e decisões caberiam aos pais, levantando questões morais, especialmente quando não se trata de doenças, mas de melhoramentos (Otieno, 2015); e) Desigualdade social: o alto custo da tecnologia pode aumentar disparidades entre países ricos e pobres, favorecendo quem pode pagar por “melhorias genéticas” (Subica, 2023)
Consenso internacional e diretrizes atuais
Em 2015, um painel internacional concluiu que o uso clínico de edição germinativa deve ser adiado até que preocupações legais, éticas e técnicas sejam resolvidas. Em 2016, o Reino Unido autorizou o uso de CRISPR-Cas9 em embriões somente para pesquisa, não para reprodução (Ayanoğlu et al., 2020). Até 2020, 24 países proibiram legalmente a edição genética em embriões humanos, e outros 9 a restringem por diretrizes (Ishii et al., 2017, Macintosh et al., 2019). Nos EUA, o NIH (Instituto Nacional de Saúde) não financia pesquisas com edição genética em embriões humanos (National Institutes of Health, 2015).
Perspectivas futuras
As perspectivas futuras para o uso do sistema CRISPR/Cas9 e tecnologias associadas são extremamente promissoras e apontam para uma verdadeira transformação na medicina e na biotecnologia. À medida que avanços contínuos aprimoram a precisão, eficiência e segurança da edição genômica, a expectativa é que essas ferramentas se consolidem como pilares fundamentais na compreensão dos mecanismos moleculares das doenças, na identificação de alvos terapêuticos e na implementação de estratégias personalizadas de tratamento (Tyumentseva et al., 2023). A integração do CRISPR/Cas9 à medicina de precisão poderá permitir intervenções altamente específicas e individualizadas, com potencial para corrigir mutações genéticas causadoras de doenças ainda na fase embrionária ou pré-sintomática (Behr et al.,2021).
Além disso, a criação rápida e precisa de modelos animais utilizando CRISPR/Cas9 continuará a impulsionar o desenvolvimento de novos fármacos e terapias inovadoras, ao mesmo tempo em que possibilita testes pré-clínicos mais eficientes e representativos. Tecnologias derivadas ou complementares, como Cas12, Cas13, Cas14, ampliam ainda mais as possibilidades de atuação, oferecendo alternativas para edição de RNA, atuação em regiões sem sequência PAM e utilização em contextos celulares mais desafiadores. Embora desafios como efeitos off-target, entrega eficiente dos componentes e regulação ética e legal ainda existam, os esforços globais em pesquisa e desenvolvimento estão constantemente buscando superar essas barreiras (Van Haasteren et al., 2020; Rees et al., 2018). Com base no ritmo atual de progresso, é plausível imaginar um futuro em que as ferramentas CRISPR estejam plenamente integradas à prática clínica, beneficiando pacientes com doenças genéticas, câncer, infecções virais e distúrbios complexos, e contribuindo significativamente para a promoção da saúde global.
Conclusão
A terapia gênica e a edição genômica representam uma revolução no tratamento de doenças humanas, abrindo caminho para intervenções mais precisas, duradouras e potencialmente curativas, especialmente em condições de base genética como as doenças cardiovasculares. A possibilidade de restaurar a homeostase cardíaca sem a dependência contínua de medicamentos sinaliza uma nova era terapêutica, em que transtornos antes considerados intratáveis – como certas dislipidemias, hipertensão resistente e cardiopatias hereditárias – passam a ter alternativas reais de controle ou até mesmo de cura.
Entre as tecnologias promissoras, destaca-se o uso de vetores virais, como os AAV, capazes de entregar material genético terapêutico diretamente às células cardíacas com alta especificidade e relativa segurança. Ainda assim, a implementação clínica em larga escala exige cautela. É imprescindível superar desafios relacionados à eficácia, segurança a longo prazo, custo e acesso equitativo. Ensaios clínicos robustos e regulatórios rígidos são fundamentais para assegurar que esses avanços científicos se traduzam em benefícios reais à saúde pública.
Portanto, à medida que a pesquisa e o desenvolvimento tecnológico evoluem, torna-se essencial fomentar a colaboração entre cientistas, profissionais de saúde, instituições reguladoras e pacientes. Somente com esse esforço conjunto será possível concretizar todo o potencial dessas terapias inovadoras, promovendo não apenas o prolongamento da vida, mas também a melhoria significativa da qualidade de vida dos indivíduos afetados por doenças cardiovasculares e outras condições genéticas complexas.
REFERÊNCIAS
AARTSMA-RUS, A. et al. Theoretical applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human Mutation, v. 30, n. 3, p. 293–299, 2009.
ABDUL-RAHMAN, T. et al. Lipid Lowering Therapy: An Era Beyond Statins. Current Problems in Cardiology, v. 47, n. 12, p. 101342, 2022. Disponível em: https://doi.org/10.1016/j.cpcardiol.2022.101342.
AKINRINADE, O. et al. Relevance of Titin missense and non-frameshifting insertions/deletions variants in dilated cardiomyopathy. Scientific Reports, [S.l.], v. 9, n. 1, p. 4093, 11 mar. 2019. DOI: https://doi.org/10.1038/s41598-01939911-x. Erratum em: Scientific Reports, v. 10, n. 1, p. 17264, 9 out. 2020.
ANDO, Y. et al. A decreased expression of angiopoietin-like 3 is protective against atherosclerosis in apoE-deficient mice. Journal of Lipid Research, [S.l.], v. 44, n. 6, p. 1216–1223, 2003.
AYANOĞLU, F. B. et al. Bioethical issues in genome editing by CRISPR-Cas9 technology. Turkish Journal of Biology, v. 44, n. 2, p. 110-120, 2020. Disponível em: https://doi.org/10.3906/biy-1912-52.
BAK, R. O.; GOMEZ-OSPINA, N.; PORTEUS, M. H. Gene editing on center stage. Trends in Genetics, v. 34, n. 8, p. 600–611, 2018. Disponível em: https://doi.org/10.1016/j.tig.2018.05.004.
BAK, R. O.; PORTEUS, M. H. CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Reports, v. 20, p. 750-756, 2017.
BAO, W. et al. The relation of parental cardiovascular disease to risk factors in children and young adults. Circulation, v. 91, n. 2, p. 365-71, 1995. Disponível em: https://doi.org/10.1161/01.cir.91.2.365.
BEHR, M. et al. Avoiding the off-target effects of CRISPR/Cas9 system is still a challenging accomplishment for genetic transformation. Gene, v. 700, p. 176178, 2019. Disponível em: https://doi.org/10.1016/j.gene.2019.03.019.
BÉROUD, C. et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Human Mutation, v. 28, n. 2, p. 196–202, 2007.
BOEKHOLDT, S. M. et al. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta-analysis of statin trials. Journal of the American College of Cardiology, v. 64, n. 5, p. 485-94, 2014. Disponível em: https://doi.org/10.1016/j.jacc.2014.02.615.
BONOWICZ, K. et al. CRISPR-Cas9 in Cardiovascular Medicine: Unlocking New Potential for Treatment. Cells, v. 14, n. 2, p. 131, 2025. Disponível em: https://doi.org/10.3390/cells14020131.
BRUGADA, J. et al. Present status of Brugada syndrome: JACC state-of-the-art review. Journal of the American College of Cardiology, v. 72, p. 1046–1059, 2018. Disponível em: https://doi.org/10.1016/j.jacc.2018.06.037.
CAHILL, Thomas J.; ASHRAFIAN, Houman; WATKINS, Hugh. Genetic cardiomyopathies causing heart failure. Circulation Research, v. 113, n. 6, p. 660–675, 2013. DOI: https://doi.org/10.1161/CIRCRESAHA.113.300282.
CHENG, X. et al. Positional cloning of quantitative trait nucleotides for blood pressure and cardiac QT-interval by targeted CRISPR/Cas9 editing of a novel long non-coding RNA. PLoS Genetics, v. 13, n. 8, p. e1006961, 2017. Disponível em: https://doi.org/10.1371/journal.pgen.1006961.
CHONG, B. et al. Global burden of cardiovascular diseases: projections from 2025 to 2050. European Journal of Preventive Cardiology, 2024. Disponível em: https://doi.org/10.1093/eurjpc/zwae281.
COHEN, J. C. et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. New England Journal of Medicine, v. 354, n. 12, p. 1264–72, 2006. Disponível em: https://doi.org/10.1056/NEJMoa054013.
DE GRAEFF, N. et al. The ethics of genome editing in non-human animals: a systematic review of reasons reported in the academic literature. Philosophical Transactions of the Royal Society B , v. 374, n. 1772, p. 20180106, 2019. Disponível em: https://doi.org/10.1098/rstb.2018.0106.
DI CEASARE, M. et al. The Heart of the World. Global Heart, v. 19, n. 1, p. 11, 2024. Disponível em: https://doi.org/10.5334/gh.1288.
DONOHOUE, P. D. et al. Advances in Industrial Biotechnology Using CRISPRCas Systems. Trends in Biotechnology, v. 36, n. 2, p. 134-146, 2018. Disponível em: https://doi.org/10.1016/j.tibtech.2017.07.007.
DORANS, K. S. et al. Trends in prevalence and control of hypertension according to the 2017 American College of Cardiology/American Heart Association (ACC/AHA) guideline. Journal of the American Heart Association, v. 7, p. e008888, 2018. Disponível em: 10.1161/JAHA.118.008888
DOUDNA, J. A.; CHARPENTIER, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science, v. 346, p. 1258096, 2014.
DUARDO-SANCHEZ, A. CRISPR-Cas in medicinal chemistry: applications and regulatory concerns. Current Topics in Medicinal Chemistry, v. 17, p. 3308, 2017. Disponível em: https://doi.org/10.2174/1568026618666171211151142.
FITZGERALD, K. et al. A highly durable RNAi therapeutic inhibitor of PCSK9. New England Journal of Medicine, v. 376, p. 41–51, 2017. Disponível em: https://doi.org/10.1056/NEJMoa1609243.
FITZGERALD, K. et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet, v. 383, p. 60–68, 2014. Disponível em: https://doi.org/10.1016/s0140-6736(13)61914-5.
FOSS-FREITAS, M. C. et al. Selective targeting of angiopoietin-like 3 (ANGPTL3) with vupanorsen for the treatment of patients with familial partial lipodystrophy (FPLD): results of a proof-of-concept study. Lipids in Health and Disease, v. 20, n. 1, p. 174, 2021.
GAUDET, D. et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridemia. European Heart Journal, v. 41, n. 40, p. 3936–45, 2020.
GOLDENBERG, I.; MOSS, A. J. Long QT syndrome. Journal of the American College of Cardiology, v. 51, p. 2291–300, 2008. Disponível em: https://doi.org/10.1016/j.jacc.2008.02.068.
HERAI, R. H. Avoiding the off-target effects of CRISPR/Cas9 system is still a challenging accomplishment for genetic transformation. Gene, v. 700, p. 176178, 2019. Disponível em: https://doi.org/10.1016/j.gene.2019.03.019.
HOOPER, A. J. et al. Verve-101, a CRISPR-based editing therapy designed to permanently inactivate hepatic PCSK9 and reduce LDL-cholesterol. Expert Opinion on Investigational Drugs, v. 33, n. 8, p. 753–6, 2024. Disponível em: https://doi.org/10.1080/13543784.2024.2369747.
HORIE, T. et al. VERVE-101: a promising CRISPR-based gene editing therapy that reduces LDL-C and PCSK9 levels in HeFH patients. European Heart Journal – Cardiovascular Pharmacotherapy , v. 10, n. 2, p. 89–90, 2024. Disponível em: https://doi.org/10.1093/ehjcvp/pvad103.
HORIE, Takahiro; ONO, Koh. VERVE-101: a promising CRISPR-based gene editing therapy that reduces LDL-C and PCSK9 levels in HeFH patients. European Heart Journal – Cardiovascular Pharmacotherapy, v. 10, n. 2, p. 89– 90, mar. 2024. Disponível em: https://doi.org/10.1093/ehjcvp/pvad103.
ISHII, T. Germ line genome editing in clinics: the approaches, objectives and global society. Briefings in Functional Genomics, v. 16, p. 46, 2017. Disponível em: https://doi.org/10.1093/bfgp/elv053.
JOHANSEN, A. K. et al. Postnatal Cardiac Gene Editing Using CRISPR/Cas9 With AAV9-Mediated Delivery of Short Guide RNAs Results in Mosaic Gene Disruption. Circulation Research, v. 121, n. 10, p. 1168-1181, 2017. Disponível em: https://doi.org/10.1161/CIRCRESAHA.116.310370.
KASSIM, S. H. et al. Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS One, v. 5, n. 10, p. e13424, 2010. Disponível em https://doi.org/10.1371/journal.pone.0013424.
KERSTEN, S. ANGPTL3 as therapeutic target. Current Opinion in Lipidology , v. 32, n. 6, p. 335–41, 2021.Disponível em 10.1097/MOL.0000000000000789
KOBLAN, L. W. et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature , v. 589, p. 608–614, 2021. Disponível em: https://doi.org/10.1038/s41586-021-03217-9.
KOENIG, M. et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. American Journal of Human Genetics, v. 45, n. 4, p. 498–506, 1989.
LEE, R. G. et al. Efficacy and Safety of an Investigational Single-Course
CRISPR Base-Editing Therapy Targeting PCSK9 in Nonhuman Primate and Mouse Models. Circulation, v. 147, n. 3, p. 242-253, 2023. Disponível em: https://doi.org/10.1161/CIRCULATIONAHA.122.062132.
LI, J. et al. Therapeutic Exon Skipping Through a CRISPR-Guided Cytidine Deaminase Rescues Dystrophic Cardiomyopathy in Vivo. Circulation, v. 144, n. 22, p. 1760-1776, 2021. Disponível em: https://doi.org/10.1161/CIRCULATIONAHA.121.054628.
LI, Q. et al. The distribution of cardiovascular-related comorbidities in different adult-onset cancers and related risk factors: analysis of 10 year retrospective data. Frontiers in Cardiovascular Medicine, Lausanne, v. 8, 14 set. 2021. Art. 695454. Disponível em: https://doi.org/10.3389/fcvm.2021.695454.
LI, Z. H.; WANG, J.; XU, J. P. Recent advances in CRISPR-based genome editing technology and its applications in cardiovascular research. Military Medical Research , v. 10, p. 12, 2023. Disponível em: https://doi.org/10.1186/s40779-023-00447-x.
LIU, W. et al. Applications and challenges of CRISPR-Cas gene-editing to disease treatment in clinics. Precision Clinical Medicine, v. 4, n. 3, p. 179-191, 2021. Disponível em: https://doi.org/10.1093/pcmedi/pbab014.
MACINTOSH, K. L. Heritable genome editing and the downsides of a global moratorium. The CRISPR Journal, v. 2, p. 272, 2019. Disponível em: https://doi.org/10.1089/crispr.2019.0016.
MALI, P. et al. RNA-guided human genome engineering via Cas9. Science, v. 339, n. 6121, p. 823-6, 2013. Disponível em: https://doi.org/10.1126/science.1232033.
MAO, Q.; KONG, Y. Global burden of cardiovascular diseases attributable to diet low in vegetables from 1990 to 2021 and forecasting the future trends: a population-based study. Frontiers in Cardiovascular Medicine, v. 11, art. 1491869. Disponível em: https://doi.org/10.3389/fcvm.2024.1491869.
MCAFEE, Q. et al. Truncated titin proteins in dilated cardiomyopathy. Science Translational Medicine, v. 13, n. 618, p. eabd7287, 2021. Disponível em: https://doi.org/10.1126/scitranslmed.abd7287.
NAWROCKI, J. W. et al. Reduction of LDL cholesterol by 25% to 60% in patients with primary hypercholesterolemia by atorvastatin, a new HMG-CoA reductase inhibitor. Arteriosclerosis, Thrombosis, and Vascular Biology, v. 15, n. 5, p. 678–682, 1995.Disponível em: 10.1161/01.atv.15.5.678
NEWMAN, C. B. et al. Statin Safety and Associated Adverse Events: A Scientific Statement from the American Heart Association. Arteriosclerosis, Thrombosis, and Vascular Biology, v. 39, n. 2, p. e38–81, 2019. Disponível em: 10.1161/ATV.0000000000000073
OOSTVEEN, R. F. et al. New approaches for targeting PCSK9: small-interfering ribonucleic acid and genome editing. Arteriosclerosis, Thrombosis, and Vascular Biology, v. 43, n. 7, p. 1081–1092, 2023. Disponível em: https://doi.org/10.1161/ATVBAHA.122.317963.
OTIENO, M. O. CRISPR-Cas9 human genome editing: challenges, ethical concerns and implications. Journal of Clinical Research and Bioethics, v. 6, p. 253, 2015. Disponível em https://doi.org/10.4172/2155-9627.1000253.
OUDE BLENKE, E. et al. CRISPR-Cas9 Gene Editing: Delivery Aspects and Therapeutic Potential. Journal of Controlled Release, v. 244, p. 139–148, 2016. Disponível em: 10.1016/j.jconrel.2016.08.002.
PATEL, P. N. et al. Contribution of Noncanonical Splice Variants to TTN Truncating Variant Cardiomyopathy. Circulation: Genomic and Precision Medicine, v. 14, n. 5, p. e003389, 2021. Disponível em: https://doi.org/10.1161/CIRCGEN.121.003389.
PUCCINI, M.; LANDMESSER, U.; RAUCH, U. Pleiotropic effects of PCSK9: focus on thrombosis and haemostasis. Metabolites, v. 12, n. 3, p. 226, 2022. Disponível em: https://doi.org/10.3390/metabo12030226.
REES, H. A.; LIU, D. R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nature Reviews Genetics, v. 19, p. 770–788, 2018. Disponível em: https://doi.org/10.1038/s41576-018-0059-1.
REICHART, D. et al. Efficient in vivo genome editing prevents hypertrophic cardiomyopathy in mice. Nature Medicine, v. 29, p. 412–421, 2023. Disponível em: https://doi.org/10.1038/s41591-022-02190-7.
ROH, D. S. et al. CRISPR Craft: DNA editing the reconstructive ladder. Plastic and Reconstructive Surgery, v. 142, p. 1355, 2018. Disponível em: https://doi.org/10.1097/PRS.0000000000004863
SABATINE, M. S. et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. New England Journal of Medicine, v. 376, n. 18, p. 1713–22, 2017. Disponível em: https://doi.org/10.1056/NEJMoa1615664.
SCHWARTZ, G. G. et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. New England Journal of Medicine, v. 379, n. 22, p. 20972107, 2018. Disponível em: https://doi.org/10.1056/NEJMoa1801174.
SHINWARI, Z. K. et al. Ethical issues regarding CRISPR-mediated genome editing. Current Issues in Molecular Biology, v. 26, p. 103, 2017. Disponível em: https://doi.org/10.21775/cimb.026.103.
SONG, Y. et al. Deciphering common Long QT syndrome using CRISPR/Cas9 in human-induced pluripotent stem cell-derived cardiomyocytes. Frontiers in Cardiovascular Medicine, v. 9, 2022. Disponível em https://doi.org/10.3389/fcvm.2022.889519
STATEMENT ON NIH FUNDING OF RESEARCH USING GENE-EDITING TECHNOLOGIES IN HUMAN EMBRYOS. 2015.
SUBICA, A. M. CRISPR in public health: the health equity implications and role of community in gene-editing research and applications. American Journal of Public Health, v. 113, n. 8, p. 874–882, ago. 2023. DOI: https://doi.org/10.2105/AJPH.2023.307315.
SUN, H. et al. CRISPR/Cas9 mediated deletion of the angiotensinogen gene reduces hypertension: a potential for cure? Hypertension, v. 77, n. 6, p. 1990– 2000, 2021. Disponível em: https://doi.org/10.1161/HYPERTENSIONAHA.120.16870.
TESTER, D. J.; ACKERMAN, M. J. Genetics of long QT syndrome. Methodist DeBakey Cardiovascular Journal, v. 10, n. 1, p. 29–33, 2014. Disponível em: https://doi.org/10.14797/mdcj-10-1-29.
TIRRONEN, A. et al. Recent advances in novel therapies for lipid disorders. Human Molecular Genetics, v. 28, n. R1, p. R49–54, 2019. Disponível em: 10.1093/hmg/ddz132
TOEPFER, C. N. et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Science Translational Medicine, v. 11, p. eaat1199, 2019. Disponível em: https://doi.org/10.1126/scitranslmed.aat1199.
TONELLI, M. et al. Efficacy of statins for primary prevention in people at low cardiovascular risk: a meta-analysis. CMAJ, v. 183, n. 16, p. E1189-E1202, 2011. Disponível em: https://doi.org/10.1503/cmaj.101280.
TONTONOZ, P. et al. Long noncoding RNA facilitated gene therapy reduces atherosclerosis in a murine model of familial hypercholesterolemia. Circulation, v. 136, p. 776–8, 2017. Disponível em: 10.1161/CIRCULATIONAHA.117.029002
TYUMENTSEVA, M. et al. CRISPR/Cas9 Landscape: Current State and Future Perspectives. International Journal of Molecular Sciences, v. 24, n. 22, p. 16077, 2023. Disponível em: https://doi.org/10.3390/ijms242216077.
UDDIN, F. et al. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Frontiers in Oncology, v. 10, p. 1387, 2020. Disponível em: https://doi.org/10.3389/fonc.2020.01387.
VAN HAASTEREN, J. et al. The delivery challenge: fulfilling the promise of therapeutic genome editing. Nature Biotechnology, v. 38, p. 845–855, 2020. Disponível em: https://doi.org/10.1038/s41587-020-0565-5.
VERVE THERAPEUTICS. Home. 2025. Disponível em: https://ir.vervetx.com/news-releases/news-release-details/verve-therapeuticsannounces-updates-its-pcsk9-program. Acesso em: 25 de abril de 2025.
VUTTHIKRAIVIT, W. et al. Worldwide Prevalence of Brugada Syndrome: A Systematic Review and Meta-Analysis. Acta Cardiologica Sinica, v. 34, n. 3, p. 267-277, 2018. Disponível em: https://doi.org/10.6515/ACS.201805_34(3).20180302B.
WAGHULDE, H. et al. Attenuation of microbiota dysbiosis and hypertension in a CRISPR/Cas9 gene ablation rat model of GPER1. Hypertension, v. 72, p. 1125– 32, 2018. Disponível em: https://doi.org/10.1161/HYPERTENSIONAHA.118.11175.
WILCOX, J. E.; HERSHBERGER, R. E. Genetic cardiomyopathies. Current Opinion in Cardiology, v. 33, n. 3, p. 354-362, 2018. Disponível em: https://doi.org/10.1097/HCO.0000000000000512.
WORLD HEALTH ORGANIZATION. Cardiovascular diseases (CVDs). Disponível em: https://www.who.int/news-room/factsheets/detail/cardiovascular-diseases-(cvds). Acesso em: 22 abr. 2025.
WU, S. et al. Base editing effectively prevents early-onset severe cardiomyopathy in Mybpc3 mutant mice. Cell Research, v. 34, p. 327–330, 2024. Disponível em: https://doi.org/10.1038/s41422-024-00930-7.
WU, S. et al. Base editing effectively prevents early-onset severe cardiomyopathy in Mybpc3 mutant mice. Cell Research, v. 34, p. 327–330, 2024. Disponível em: https://doi.org/10.1038/s41422-024-00930-7.
YIN, J. et al. Optimizing genome editing strategy by primer-extension-mediated sequencing. Cell Discovery, v. 5, p. 18, 2019. Disponível em: https://doi.org/10.1038/s41421-019-0088-8.
ZHANG, H. et al. AAV-mediated gene therapy: advancing cardiovascular disease treatment. Frontiers in Cardiovascular Medicine, v. 9, 2022. Disponível em: https://doi.org/10.3389/fcvm.2022.952755
ZHONG, R. et al. A Preclinical Study on Brugada Syndrome with a CACNB2 Variant Using Human Cardiomyocytes from Induced Pluripotent Stem Cells. International Journal of Molecular Sciences, v. 23, n. 15, p. 8313, 2022. Disponível em: https://doi.org/10.3390/ijms23158313
1E-mail: nemenahhh@gmail.com CRM/PR 40.185
Residente em Clínica Médica Hospital Memorial Uningá
2E-mail: zeliofedattojr@yahoo.com.br CRM/PR 39.013
Coordenador do Curso de Medicina Uningá
Diretor Médico das Unidades Hospitalar Memorial I e Ambualtorial II