CHALLENGES IN THE DIAGNOSIS OF MICROSCOPIC POLYANGIITIS: A CASE REPORT
DESAFÍOS EN EL DIAGNÓSTICO DE LA POLIANGIITIS MICROSCÓPICA: UN CASO CLÍNICO
REGISTRO DOI: 10.69849/revistaft/ni10202506081442
Najla Neme Dutra1
Jerdal Micael Quilla Morsoletto2
Francismar Prestes Leal3
Gabriel Nascimento de Oliveira4
Zelio Fedatto Junior5
RESUMO
Introdução: A poliangeíte microscópica (PAM) é uma vasculite rara e potencialmente fatal que afeta pequenos vasos, geralmente associada à positividade de anticorpos anticitoplasma de neutrófilos (ANCA). Seus sintomas podem ser bastante inespecíficos, o que retarda a identificação da doença e o início dos tratamentos adequados. Objetivo: Este estudo tem como objetivo relatar e analisar um caso complexo de PAM, com foco nos desafios diagnósticos e terapêuticos enfrentados. Metodologia: trata-se de estudo de caso de um paciente de 51 anos, inicialmente atendido com uveíte e lesões cutâneas, que evoluiu com glomerulonefrite rapidamente progressiva e complicações pulmonares graves. Os dados foram coletados por meio de análise documental do prontuário clínico, exames laboratoriais e biópsias, e foram complementados com entrevistas da equipe assistente. Resultados: O paciente apresentou quadro clínico variado e inespecífico, dificultando o diagnóstico precoce. A confirmação ocorreu tardiamente por meio de biópsia renal e marcadores imunológicos, com progressão para insuficiência renal e hemorragia alveolar. Mesmo com o uso de pulsoterapia e hemodiálise, o paciente evoluiu a óbito. Conclusão: O estudo evidencia a importância da suspeição clínica precoce e da abordagem multidisciplinar na PAM. A diversidade de manifestações e a falta de achados específicos em fases iniciais reforçam a necessidade de estratégias diagnósticas sistematizadas para melhorar o prognóstico.
Palavras-chave: Poliangeíte Microscópica, Diagnóstico Clínico, Morbimortalidade, Relato de caso.
ABSTRACT
Introduction: Microscopic polyangiitis (MPA) is a rare and potentially fatal vasculitis that affects small vessels, usually associated with neutrophil cytoplasmic antibodies (ANCA) positivity. Its symptoms can be quite nonspecific, which delays the identification of the disease and the initiation of appropriate treatments. Objective: This study aims to report and analyze a complex case of MPA, focusing on the diagnostic and therapeutic challenges faced. Methodology: This is a case study of a 51-year-old patient, initially treated for uveitis and skin lesions, who developed rapidly progressive glomerulonephritis and severe pulmonary complications. Data were collected through documentary analysis of medical records, laboratory tests, and biopsies, and were supplemented with interviews with the attending team. Results: The patient presented with a varied and nonspecific clinical picture, making early diagnosis difficult. Confirmation occurred at a late stage through renal biopsy and immunological markers, with progression to renal failure and alveolar hemorrhage. Despite treatment with pulse therapy and hemodialysis, the patient died. Conclusion: This study highlights the importance of early clinical suspicion and a multidisciplinary approach in MPA. The diversity of manifestations and the lack of specific findings in early stages reinforce the need for systematic diagnostic strategies to improve patient outcomes.
KEYWORDS: Microscopic Polyangiitis, Clinical Diagnosis, Morbidity, Mortality, Case report.
1. INTRODUÇÃO
A poliangeíte microscópica (PAM) é uma doença inflamatória rara que afeta pequenos vasos sanguíneos (Li et al., 2021; Suppiah et al., 2022; Ti, 2023). A fisiopatologia da PAM envolve a ativação dos neutrófilos pelos anticorpos anticitoplasma de neutrófilos (ANCA), levando à liberação de mediadores inflamatórios e espécies reativas de oxigênio. Isso resulta em dano endotelial, inflamação e necrose da parede vascular, culminando em isquemia tecidual e disfunção orgânica (Li et al., 2021; Suppiah et al., 2022).
A PAM afeta múltiplos sistemas com manifestações clínicas variadas, o que pode dificultar o diagnóstico precoce e adequado (Li et al., 2021; Suppiah et al., 2022; Ti, 2023). Nos sinais e sintomas sistêmicos, os pacientes podem apresentar febre, artralgia, perda de peso e algumas manifestações cutâneas devido ao comprometimento dos pequenos vasos, como erupções cutâneas, papulosas e lesões necrosantes (Katerenchuk et al., 2021; Tobiáš et al., 2020). Em casos mais atípicos, a PAM pode resultar em sintomas gastrointestinais, cardíacos, neurológicos, hematológicos e oftalmológicos (Arshad et al., 2022; Caster et al., 1996; Lee, 2020; Moosig, 2008; Rivera et al., 2020).
Entre os órgãos mais frequentemente afetados pela PAM, os rins ocupam papel central, com quase todos os pacientes apresentando algum grau de glomerulonefrite rapidamente progressiva. O comprometimento renal se expressa por hematúria, proteinúria e cilindros hemáticos, podendo ser silencioso até estágios avançados (Pagnoux, 2016; Li et al., 2021; Suppiah, et al., 2022). Os pulmões também são frequentemente envolvidos, podendo manifestarse por tosse, dispneia, hemoptose significativa, hemorragia alveolar, fibrose pulmonar ou doença pulmonar intersticial. Além disso, são descritas manifestações como púrpura, mononeurite múltipla e vasculite mesentérica (Song et al. 2024; Yang, et al., 2017; Zhang, et al., 2024). As lesões cutâneas — como púrpura palpável, livedo reticular, úlceras e nódulos — podem estar presentes em até 60% dos casos, por vezes como manifestação inicial (De Sousa, et al., 2019; Mukhtyar, et al, 2023; Sunderkötter et al. 2018). Todas essas manifestações inespecíficas e sua apresentação clínica variada representam desafios no diagnóstico da PAM, acarretando diagnóstico tardio, já que os sintomas podem trazer fatores de confusão com outras doenças (Ti, 2023; Tobiáš et al., 2020), reforçando a necessidade de uma abordagem diagnóstica abrangente e sistematizada (Katerenchuk et al., 2021; Ti, 2023)
A incidência global da PAM varia entre 2,7 e 94 casos por milhão de habitantes, com predominância em países do sul da Europa, como Itália e Espanha, e em nações asiáticas, como Japão e China (Watts, et al., 2015). Nessas regiões, a PAM é mais comum que a granulomatose com poliangiite (GPA), enquanto no norte da Europa e em populações de ascendência europeia, a GPA é mais prevalente (Watts et al., 2015).
No Brasil, a doença afeta cerca de 400 pessoas a cada 1 milhão (Ministério Da Saúde, 2023). Além disso, a epidemiologia das vasculites sistêmicas no país, incluindo a PAM, apresenta particularidades regionais. Um estudo realizado em centros especializados das regiões Sudeste e Nordeste identificou que a doença de Behçet e a arterite de Takayasu são as vasculites mais frequentes. A PAM representou 3,4% dos casos na região Sudeste (Souza et al., 2018).
A idade média de início da PAM situa-se entre 50 e 60 anos, com um aumento na incidência em faixas etárias mais avançadas (Watts et al., 2015). Embora alguns estudos apontem uma leve predominância masculina, com uma razão homem:mulher de aproximadamente 1,8:1, outros relatam uma distribuição equitativa entre os sexos (Watts et al., 2015). No Brasil, a ocorrência é mais observada no sexo feminino com idade média de diagnóstico por volta de 47,6 anos para a PAM (Ministério Da Saúde, 2023).
A mortalidade da PAM é mais elevada nos primeiros meses após o diagnóstico, sendo a vasculite responsável por 32% a 50% dos óbitos registrados (Corral-Gudino et al., 2011). Fatores como idade avançada, comprometimento renal e complicações decorrentes do tratamento, especialmente infecções, estão diretamente relacionadas ao aumento do risco de mortalidade (Manabe et al., 2024; Nguyen et al., 2020).
O diagnóstico precoce da doença é fundamental para iniciar o tratamento com imunossupressão adequada, que pode significar um melhor prognóstico e qualidade de vida ao paciente (Katerenchuk et al., 2021; Ti, 2023). Dados epidemiológicos da doença no Brasil são escassos, porém uma revisão de literatura sobre vasculites secundárias à PAM indica que a doença é rara e frequentemente diagnosticada de forma tardia, afetando negativamente o prognóstico dos pacientes (De Azevedo et al., 2018).
A complexidade da doença, sua raridade, seus sinais e sintomas diversos, tornam o diagnóstico dessa doença um importante desafio para a saúde pública. Por isso, buscando contribuir com o conhecimento da comunidade médica, o objetivo deste estudo é relatar um caso de um paciente com PAM, descrevendo e analisando seus aspectos diagnósticos, terapêuticos e prognósticos, oferecendo insights que possam auxiliar no manejo de situações semelhantes.
2. METODOLOGIA
2.1. TIPO DE ESTUDO E ASPECTOS ÉTICOS
Trata-se de estudo de caso único, do tipo retrospectivo, observacional e descritivo, utilizando dados de um paciente com suspeita de PAM atendido no Hospital Memorial Uningá e no Hospital Santa Casa, ambos localizados na cidade de Maringá – Paraná. O sujeito de estudo foi selecionado por conveniência, conforme atendimento clínico já em curso, observado e acompanhado em ambiente intra-hospitalar (Enfermaria e unidade de terapia intensiva (UTI) de ambas as instituições) e extra-hospitalar (Unidade II Memorial Uningá – Ambulatório de especialidades). O estudo iniciou após assinatura do termo de consentimento livre e esclarecido (TCLE), assinado pelo responsável legal – neste caso, esposa do paciente. O TCLE foi aplicado presencialmente, em local reservado, com explicação detalhada sobre os objetivos, riscos e benefícios da pesquisa. O estudo foi aprovado pelo Comitê de Ética do Centro Universitário UNINGÁ, (CAAE: 87978925.0.0000.5220).
2.2. COLETA E ANÁLISE DOS DADOS
Os dados foram coletados por profissionais vinculados às instituições, através de prontuário eletrônico (Plataforma Tasy) e entrevistas com a equipe assistencial, respeitando os princípios éticos e os direitos do sujeito da pesquisa. As informações coletadas foram: histórico clínico; exames laboratoriais; exames de imagem; e biopsias realizadas (anatomopatológico e imunofluorescência). Após a coleta, foram organizados de forma cronológica para melhor compreensão. A análise realizada foi qualitativa e descritiva, com base na interpretação dos dados clínicos, laboratoriais, terapêuticos e contextuais obtidos.
3. RELATO DE CASO
Paciente identificado como “F. O. T. C.”, sexo masculino, 51 anos, proveniente do Peru, residente da cidade de Marialva – Paraná, com esposa e dois filhos, hígido e sem comorbidades prévias. Procurou atendimento médico por conta própria, com queixa de surgimento súbito de escotomas flutuantes em olho esquerdo, evoluindo com perda gradual da acuidade visual (padrão centro-periférico). Nesta ocasião, um especialista em retina avaliou e indicou procedimento de fotocoagulação a laser e infiltração intravítrea de angiogênico em olho esquerdo, devido a um possível diagnóstico de uveíte/endoftalmite a esclarecer.
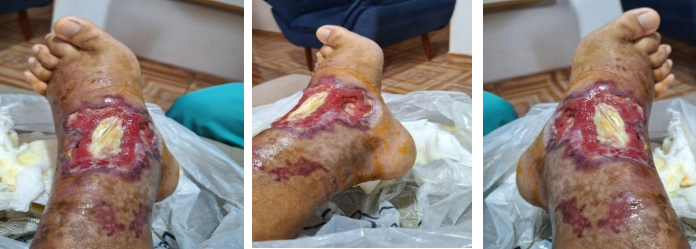
Nos dias que seguiram, o paciente iniciou quadro de dor insidiosa e edema leve em região de pé e tornozelo. Evoluiu para perda progressiva da acuidade visual em olho esquerdo, com queixa concomitante de surgimento de lesão bolhosa em região de dorso do pé direito, associado à presença de sinais flogísticos e parestesia local. Após cerca de duas semanas do primeiro atendimento, apresentou perda total da visão no olho esquerdo, sem alterações na acuidade visual do olho direito e com piora gradual da lesão bolhosa (Figura 1). O paciente foi reavaliado e observada a piora significativa de quadro clinico, foi realizada a internação em Unidade de Pronto Atendimento (UPA) em Marialva, para investigação e tratamento adequado. O paciente foi então transferido para o Hospital Memorial UNINGA, com suspeita diagnóstica de erisipela bolhosa em membro inferior direito. Foram realizados exames laboratoriais de admissão, com provas proinflamatórias altas (PCR 138 mg/dl e VHS 101mm), bem como leucocitose moderada associada, sem desvios em hemograma.
Figura 1. Lesões iniciais observadas no paciente, entre o começo e meio de julho de 2024.
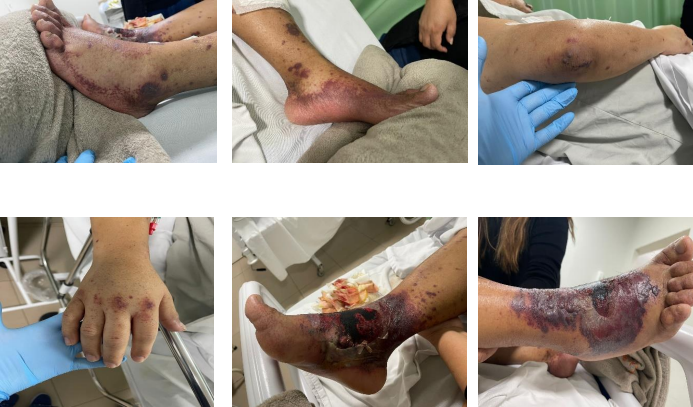
Após 24h de internação, foi observado quadro de lesões sugestivas purpúricas sobrepostas ao quadro bolhoso prévio em membro inferior direito, se estendendo para sola do pé e tornozelo ipsilateral. Foi optado então pela realização de biopsia de pele, que retornou com resultado de epiderme sem atipias, presença de derme com infiltrado leucocitário predominantemente eosinofílico, com alguns neutrófilos e ausência de granulomas, trombos e/ou fungos nas amostras analisadas. Houve também a realização de ultrassom doppler venoso, para excluir evento trombótico periférico, que retornou com resultado negativo.
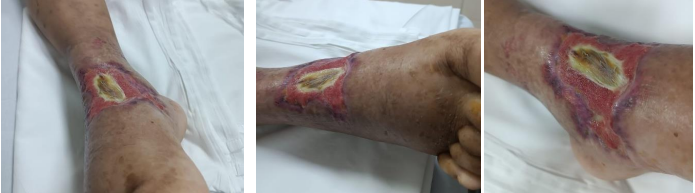
Paciente evoluiu com piora das lesões bolhosas e instabilidade hemodinâmica após 48h da admissão, e foi transferido para leito de UTI, para melhor monitoramento e estabilização de quadro clínico. Neste momento, foi iniciada pulsoterapia com metilprednisolona (1g/dia por 5 dias) (Figura 2), com escalonamento de antibioticoterapia. Foram coletadas hemoculturas para aprofundamento da investigação com exames laboratoriais e de imagem. Exame de VHS permanecia alto (70mm), e marcadores de p-ANCA reagente. c-ANCA, FAN, C3, C4 e sorologias como hepatites, sífilis e HIV foram não reagentes. Uma tomografia de abdome evidenciou pequena quantidade de líquido livre em cavidade e tórax, com derrame pleural bilateral moderado (Figura 4).
Figura 2. Evolução das lesões após primeira pulsoterapia, final de julho de 2024.
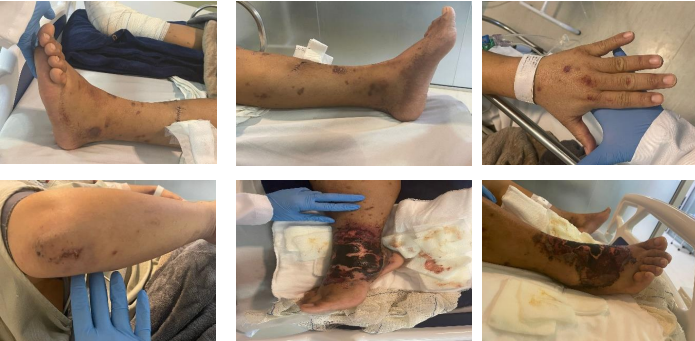
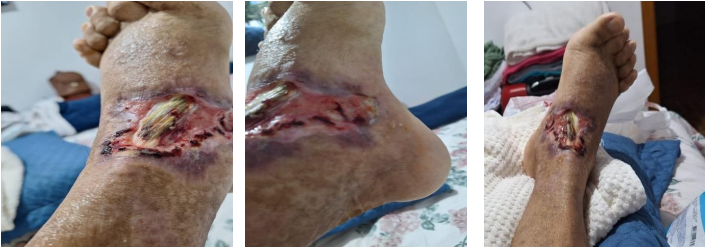
Após alguns dias de monitoramento em UTI e diferentes tratamentos, o paciente recebeu alta do setor, com retorno para a enfermaria devido à melhora clínica e restabelecimento da estabilidade hemodinâmica. Neste momento, foi realizado debridamento em membro inferior direito por cirurgião vascular, que removeu o máximo de tecido morto presente na lesão, preservando as estruturas nobres (Figura 3).
Figura 3. Evolução da lesão após desbridamento.
A. Início de Agosto, 2024 – logo após desbridamento
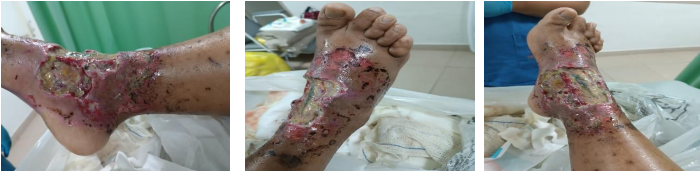
B. Metade de Agosto, 2024
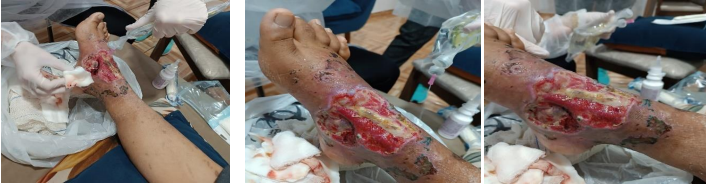
C. Início de Setembro, 2024
D. Final de Setembro, 2024
E. Início de Outubro, 2024
Após pouco mais de um mês do primeiro atendimento, paciente recebeu alta hospitalar, com acompanhamento ambulatorial para iniciar reabilitação motora e desmame de corticoide (Prednisona). Neste momento, foram checados os resultados de exames complementares, como fator reumatoide, anticorpo CCP e anti-proteinase PR3, todos não reagentes. O resultado do exame de urina mostrou leve proteinúria (2+), e hematúria microscópica (> 1mi), mantendo função renal preservada (creatinina 1,28 mg/dl e ureia 68 mg/dl), demais sem particularidades.
Dois meses após a alta hospitalar, paciente retornou ao serviço com quadro de hiporexia, náusea, emese persistente associado a astenia intensa progressiva, escurecimento de diurese com espuminuria e pouca resposta a medicamentos sintomáticos. Exames laboratoriais solicitados na ocasião indicaram aumento significativo de função renal (creatinina 3,66 mg/dl e ureia 85mg/dl), bem como, leucocitúria, hematúria macroscópica (>600 mil) e proteinúria 24h elevado (>3,5 g) em amostra de urina isolada. Paciente deu entrada novamente na UPA em Marialva, e logo foi transferido para o Hospital Memorial UNINGÁ. A avaliação inicial do nefrologista indicou a necessidade de biópsia renal, com possível terapia de substituição renal caso não houvesse melhora de função renal. O paciente foi então transferido para o Hospital Santa Casa de Maringá após uma semana de reinternação, com justificativa de serviço mais estruturado e equipe direcionada da nefrologia para procedimento e acompanhamento dele.
Figura 4. Evolução de exames de imagem do paciente ao longo do curso clínico.
A. Tomografia computadorizada – junho, 2024
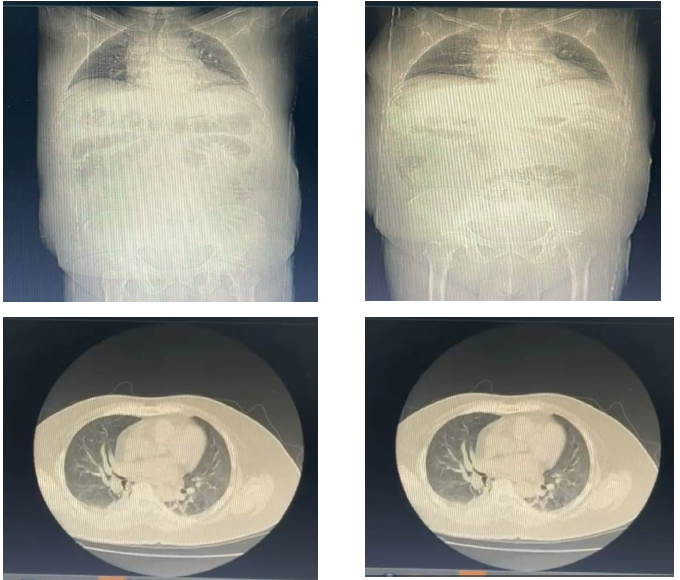
B. Exames de Raio X (à esquerda) e tomografia computadorizada (à direita) – novembro, 2024
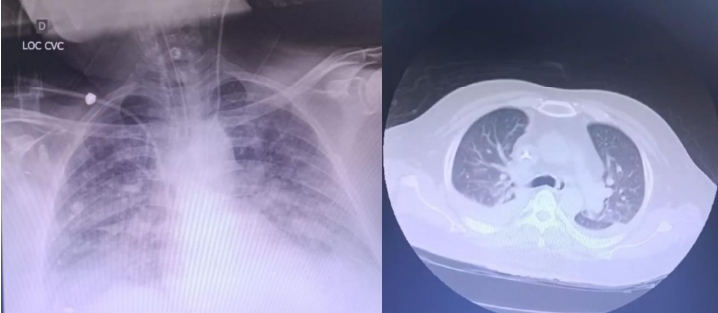
Houve persistência de insuficiência renal aguda, acrescida de glomerulopatia e hipercalemia refrataria, que apresentaram pouca resposta a corticoterapia. Por isso, dias após sua transferência interhospitalar, programaram-se duas sessões de hemodiálise, visando o controle da função renal antes da biópsia, como estratégia para identificar e iniciar o tratamento para vasculite em atividade ou nefrite o mais breve possível. Foi realizada então a biopsia em rim esquerdo, guiado por ultrassonografia, e retirada 2 peças (uma para análise histológica/anatomopatológica e outra para imunofluorescência). A anatomopatologia mostrou presença de cilindros hemáticos, infiltrados inflamatórios mistos, fibrose, lesões degenerativas difusas, cristais, tubulite e membrana basal espessada – algumas até rompidas. Já a imunofluorescência revelou presença de deposição de fibrinogênio.
O paciente também necessitou de tratamento para um episódio de infecção de urina, guiado por urocultura com boa resposta. Seguiu sem melhora de função renal e hemograma com queda abrupta de hemoglobina e hematócrito durante 05 dias sem exteriorização (HB 9.7 – 8.0 – 7.6 – 6.8 – 9.3). O padrão de imagem se manteve em tomografia de tórax e abdômen, persistindo derrame pleural bilateral, com esparsas opacidades parenquimatosas irregulares, de predomínio periférico, com focos inespecíficos associado a derrame pericárdico e líquido livre em abdome. Por persistência de sintomatologia e quadro de sobrecarga renal, pensando-se em glomerulopatia de rápida progressão e pela suspeita de atividade da vasculite de pequenos vasos (ANCA reagente – direcionado a PAM) foi iniciada nova pulsoterapia (metilprednisolona 1g/dia por 5 dias). Isso pois mesmo em hemodiálise, o paciente iniciou anasarca e oscilação pressórica.
Após cerca de um mês mantendo frequência de duas a três sessões de terapia de substituição renal semanais (conforme necessidade), o paciente realizou nova tomografia de tórax de controle. Houve mudança de padrão, com persistência de derrame pleural grau moderado, associado a múltiplos focos confluentes de atenuação em vidro fosco bilateralmente, de predomínio hilar e basal, associado a pequenas consolidações. Horas após a sessão de dialise programada para aquele dia, apresentou mal-estar súbito, emese com presença de raias de sangue (hemoptise leve) e dispneia súbita, evoluindo com instabilidade hemodinâmica. Foi então encaminhado ao pronto-atendimento para manejo de insuficiência respiratória aguda, com pouca resposta às medidas iniciais. Por conta da gravidade e refratariedade de quadro clinico, o paciente foi transferido para a UTI para monitorização.
Na madrugada do dia seguinte, paciente apresentou episódio de emese em grande quantidade, associado a intensa agitação psicomotora, seguido de quadro de hipoxia central. Foi realizada então sequência atrasada para intubação orotraqueal, e acesso central em jugular, evoluindo com parada cardiorrespiratória com ritmo cardíaco em AESP (atividade elétrica sem pulso). Sequencialmente, foram realizadas manobras de reanimação cardiopulmonar com uso de drogas preconizadas segundo ACLS (Advanced Cardiac Life Support), totalizando entre 20 e 30 minutos de manejo com retorno a circulação espontânea.
Paciente em estado gravíssimo, mantendo instabilidade hemodinâmica, brigando com ventilação mecânica e altas doses de drogas vasoativas. Foi realizado novo raio-x, solicitado para localização de acesso central, tubo e controle com persistência de opacidades pulmonares irregulares difusas bilateralmente e ausculta pulmonar com estertores difusos crepitantes em ambos hemitórax. Após algumas horas apresentou nova parada cardiorrespiratória, evoluindo a óbito.
4 DISCUSSÃO
A PAM é uma vasculite necrosante de pequenos vasos, caracterizada principalmente pela presença de glomerulonefrite pauci-imune e, frequentemente, hemorragia alveolar difusa, configurando a síndrome pulmão-rim (MSD Manual, 2023). No caso discutido, observou-se manifestações cutâneas iniciais que evoluíram com acometimento renal e pulmonar, um padrão clínico compatível com PAM. O paciente apresentava p-ANCA positivo, o que reforça o diagnóstico, visto que cerca de 60 a 80% dos casos de PAM cursam com positividade para este anticorpo (MSD Manual, 2023).
A biopsia cutânea foi realizada na região de tornozelo e dorso do pé direito, durante a fase ativa da doença, considerado o momento ideal para captação das alterações histopatológicas (Ministério da Saúde, 2023). Histologicamente, foi observado infiltrado leucocitário predominantemente de eosinófilos e neutrófilos, sem formação de granulomas e sem imunodepósitos. Contudo, a ausência de análise por imunofluorescência direta é uma limitação, pois impossibilitou a diferenciação entre vasculites pauci-imunes e aquelas mediadas por imunocomplexos, como a vasculite por IGA.
A biopsia renal, guiada por ultrassonografia, foi realizada de forma apropriada quanto à técnica, capturando regiões de córtex e medula. No entanto, é plausível que tenha ocorrido uma perda do “timing“, considerando que o paciente já apresentava síndrome nefrótica e sinais de insuficiência renal progressiva. Os achados histológicos foram discretos, revelando glomerulopatia mesangial moderada, cilindros hemáticos e discreta fibrose intersticial. A imunofluorescência demonstrou ausência de imunocomplexos, corroborando o padrão pauciimune esperado em PAM (Gomes et al., 2021).
A correlação com exames laboratoriais mostrou proteinúria moderada, hematúria acentuada e deterioração progressiva da função renal. Esses achados são característicos da glomerulonefrite rapidamente progressiva associada a PAM, sendo que a presença de cilindros hemáticos na urina reforça o envolvimento glomerular (MSD MANUAL, 2025).
Em relação à diferença entre as principais vasculites associadas a ANCA (Tabela 1), a PAM distingue-se da GPA pela ausência de granulomas necrotizantes e do acometimento predominante das vias aéreas superiores, característico da GPA. A granulomatose eosinofílica com poliangiite (GEPA) apresenta acentuada eosinofilia periférica e história de asma, elementos ausentes no paciente em questão (MSD MANUAL, 2025).
Tabela 1. Diferenças entre PAM, GPA e GEPA.


Durante a internação, o paciente evoluiu com sintomas respiratórios graves, incluindo dispneia intensa, hipoxemia e episódios de hemoptise. A tomografia de tórax evidenciou infiltrados bilaterais difusos em padrão vidro fosco, sugerindo fortemente hemorragia alveolar difusa (MSD Manual, 2023). Apesar da ausência de confirmação por broncoscopia, o conjunto clínico e radiológico é altamente sugestivo de síndrome pulmão-rim, caso comum na PMA.
A hemorragia alveolar é uma emergência médica associada a alta mortalidade se não tratada rapidamente com imunossupressão agressiva (Souza et al., 2017). O atraso no reconhecimento e no tratamento direcionado da síndrome pulmão-rim possivelmente contribuiu para a evolução fatal do paciente, que desenvolveu anasarca, insuficiência respiratória aguda e instabilidade hemodinâmica refratária.
Portanto, o conjunto de dados clínicos (lesões purpúricas, síndrome nefrótica, hemorragia alveolar), laboratoriais (p-ANCA positivo, leucocitúrio, proteinúria e hematúria) e histopatológicos (padrão pauci-imune) suporta fortemente o diagnóstico de PAM. Este caso ressalta a importância de intervenção precoce com pulsoterapia e agentes imunossupressores, como ciclofosfamida ou rituximabe para a prevenção de desfechos graves.
5 CONCLUSÃO
O caso apresentado evidencia a complexidade diagnóstica da PAM, principalmente em estágios iniciais, onde os achados histopatológicos podem ser inespecíficos e a apresentação clínica variada. A histologia não definitiva e a falta de imunofluorescência na biopsia cutânea reforçam a necessidade de uma abordagem clínica soberana, como ocorreu neste caso, onde o conjunto de sinais e sintomas guiou a hipótese diagnóstica principal. A demora na confirmação diagnóstica e no início do tratamento imunossupressor agressivo pode ter contribuído para a evolução desfavorável, culminando em complicações como hemorragia alveolar difusa, insuficiência renal e instabilidade hemodinâmica refratária.
Esse cenário reforça o papel central da avaliação clínica criteriosa como ferramenta decisiva no diagnóstico da PAM. Em contextos em que exames complementares falham em fornecer respostas conclusivas — seja por limitações técnicas, ausência de imunodepósitos em biópsias ou resultados laboratoriais inconclusivos — é o olhar atento do clínico, somado à integração das manifestações sistêmicas, que direcionam a condução do caso. A anamnese detalhada, a progressão temporal dos sintomas e o reconhecimento de padrões característicos, mesmo que sutis, tornam-se mais relevantes do que a dependência exclusiva de dados objetivos. Assim, a clínica reafirma sua soberania, especialmente em doenças raras e de alta complexidade, onde a intuição médica, baseada em experiência e conhecimento técnico, pode ser determinante para antecipação do diagnóstico e início precoce da abordagem terapêutica adequada, evitando desfechos adversos.
REFERÊNCIAS
ARSHAD, I., SITTLER, D., & , J. Microscopic Polyangiitis Presenting as Pyrexia of Unknown Origin with Neurological Symptoms and a Management Dilemma. Annals of Case Reports, n. 7, p. 844, mai./2022.
BRASIL. Ministério Da Saúde. Relatório para a sociedade: informações sobre recomendações de incorporação de medicamentos e outras tecnologias no SUS. Rituximabe para terapia de indução de remissão dos pacientes com diagnostico recente para casos de recidiva de vasculites associadas aos anticorpos ancitoplasma de neutrófilos, ativa e grave. Nº 398, Junho de 2023. Disponível em https://www.gov.br/conitec/pt-br/midias/consultas/relatorios/2023/sociedade/ReSoc398Rituximabe_VAA_final.pdf
BRASIL. Ministério da Saúde. Secretaria de Ciência, Tecnologia e Inovação e do Complexo Econômico-Industrial da Saúde. Protocolo Clínico e Diretrizes Terapêuticas: Vasculite Associada aos Anticorpos Anti-citoplasma de Neutrófilos (ANCA). Brasília, DF: Ministério da Saúde, 2024. Relatório preliminar. Disponível em: https://www.gov.br/conitec/pt-br. Acesso em: 2 maio 2025.
CASTER, J.; SHETLAR, D.; PAPPOLLA, M.; & YEE, R. Microscopic polyangiitis with ocular involvement. Archives of ophthalmology, v. 114, n. 3, p. 346-8, mar. /1996.
CORRAL-GUDINO, L.; BORAO-CENGOTITA-BENGOA, M.; DEL PINO-MONTES, J.; & LERMA-MÁRQUEZ, J. Overall survival, renal survival and relapse in patients with microscopic polyangiitis: a systematic review of current evidence. Rheumatology, v. 50, n. 8, p. 1414-23, 2011.
DE AZEVEDO, F.; LIMA, F.; DE CARVALHO, J.; DE SABOIA MONT’ALVERNE, A.; & RODRIGUES, C. Granulomatosis with polyangiitis in Northeastern Brazil: study of 25 cases and review of the literature. Advances in Rheumatology, v. 58, n. 10, p. 1-9, 2018.
DE SOUSA, E. et al. Peripheral neuropathy in ANCA-associated vasculitis: a retrospective cohort. Neurology and Therapy, v. 8, n. 1, p. 101–110, 2019.
GOMES, O. V.; ALMEIDA, B. A. D; DE SANTANA, L. F. E; RODRIGUES, M. DE S.; LOCIO, G. B. P. M.; ARAÚJO, C. S.; ROSAS, C. H. DE S.; & GUIMARÃES, M. D. Ultrasound-guided percutaneous renal biopsy at a university hospital: retrospective analysis of success and complication rates. Radiologia Brasileira, v. 54, n. 5, p. 311–317, 2021.
JENNETTE, J. C. et al. Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis & Rheumatism, v. 65, n. 1, p. 1–11, 2013.
KATERENCHUK, I.; TKACHENKO, L.; YARMOLA, T.; & TALASH, V. Microscopic polyangiitis – a view of the problem through the lens of a nephrologist. Wiadomosci lekarskie, v. 74, n. 4, p. 1024-1031, 2021.
LEE, C. S.; LEE, A. Y. Ocular manifestations of systemic vasculitis. In: BARON, M. (ed.). Vasculitis in Clinical Practice. Cham: Springer. p. 309–315, 2020.
LI, Q.; YU, L.; LI, F.; WANG, J.; CHEN, Y.; & SUN, S. The Clinical and Pathological Features of Children With Microscopic Polyangiitis. Frontiers in Pediatrics, v. 9, 2021.
MANABE, A., KADOBA, K., HIWA, R., KOTANI, T., SHOJI, M., SHIRAKASHI, M., TSUJI, H., KITAGORI, K., AKIZUKI, S., NAKASHIMA, R., YOSHIFUJI, H., YAMAMOTO, W., OKAZAKI, A., MATSUDA, S., GON, T., WATANABE, R., HASHIMOTO, M., & MORINOBU, A. Risk factors for serious infections and infection-related mortality in patients with microscopic polyangiitis: Multicentre REVEAL cohort study. Modern rheumatology., v. 34, n. 6, p. 1185–1193, 2024.
MOOSIG, F. Cardiac involvement in small- and medium-vessel vasculitides. E-Journal of Cardiology Practice, v. 6, n. 14, 2008. Disponível em: https://www.escardio.org/Journals/EJournal-of-Cardiology-Practice/Volume-6/Cardiac-involvement-in-small-and-medium-sizedvasculitides-Title-Cardiac-invo. Acesso em: 8 abr. 2025.
MSD MANUAL. Poliangeíte microscópica (PAM). [S.l.]: MSD Manual, [2023]. Disponível em: https://www.msdmanuals.com/pt/profissional/dist%C3%BArbios–dos–tecidos–conjuntivoe–musculoesquel%C3%A9tico/vasculite/poliange%C3%ADte–microsc%C3%B3pica–pam. Acesso em: 2 maio 2025.
MSD MANUAL. Síndrome pulmão-rim. [S.l.]: MSD Manual, [2023]. Disponível em: https://www.msdmanuals.com/pt/casa/dist%C3%BArbios–pulmonares–e–das–viasrespirat%C3%B3rias/doen%C3%A7as–autoimunes–dos–pulm%C3%B5es/s%C3%ADndromepulm%C3%A3o–rim?ruleredirectid=762. Acesso em: 2 maio 2025.
MUKHTYAR, C. et al. (2023). Vasculitis. In: WILSON, D. R.; FERNANDEZ, C. H. (ed.).
StatPearls. Treasure Island (FL): StatPearls Publishing. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK531484/.
NGUYEN, Y., PAGNOUX, C., KARRAS, A., QUÉMÉNEUR, T., MAURIER, F., HAMIDOU, M., QUELLEC, L., CHICHE, N., COHEN, P., RÉGENT, A., LIFERMANN, F., MÉKINIAN, A., KHOUATRA, C., HACHULLA, E., POURRAT, J., RUIVARD, M., GODMER, P., VIALLARD, J., TERRIER, B., MOUTHON, L., GUILLEVIN, L., & PUÉCHAL, X. Microscopic polyangiitis: Clinical characteristics and long-term outcomes of 378 patients from the French Vasculitis Study Group Registry. Journal of autoimmunity, 102467, 2020.
PAGNOUX, C. Updates in ANCA-associated vasculitis. European Journal of Rheumatology, v. 3, n. 3, p. 122–133, 2016.
RIVERA, A., VILLEGAS, L., LOPEZ, M., YURITZI, R., LOPEZ, C., MUÑOZ, T., AGUILAR, A., SÁNCHEZ, J., DE NAVA, J., MAGAÑA, I., CASTILLO, A., & PEREZ, A. Atypical Presentation of Microscopic Polyangiitis. Journal of the American Society of Nephrology, v. 6, n. 5, 2020.
SONG, Q.; LIU, Y.; WU, T. et al. Clinical features, radiological findings and prognosis of microscopic polyangiitis with interstitial lung disease: a retrospective matched control study. BMC Pulm Med, v. 24, n. 605, 2024.
SOUZA, A. W. S. et al. Recomendações da Sociedade Brasileira de Reumatologia para a terapia de indução para vasculite associada a ANCA. Revista Brasileira de Reumatologia, v. 57, p. S484–S496, 2017.
SOUZA, A. W. S. et al. Epidemiology of primary systemic vasculitis in a tertiary referral center in São Paulo, Brazil. Clinical and Experimental Rheumatology, v. 36, n. 111, p. 12–16, 2018.
SUNDERKÖTTER, C. et al. (2018) Cutaneous vasculitis: a review. Journal of the European Academy of Dermatology and Venereology, v. 32, n. 9, p. 1499–1513, 2018.
SUPPIAH, R.; ROBSON, J.; GRAYSON, P.; PONTE, C.; CRAVEN, A.; KHALID, S.; JUDGE, A.; HUTCHINGS, A.; MERKEL, P.; LUQMANI, R.; & WATTS, R. American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis. Annals of the Rheumatic Diseases, v. 81, p. 321 – 326, 2022.
TI, Y. (2023). Microscopic Polyangiitis is a “Masquerade Ball” in Modern Realities: A Literature Review. Open Access Journal of Urology & Nephrology, v.8, n. 4, 2023.
TOBIÁŠ, D.; BRÁZDILOVÁ, K.; KILLINGER, Z.; & PAYER, J. Microscopic polyangiitis. Vnitrni lekarstvi, v. 66, n. 4, p. 249-252, 2020.
WATTS, R. A. et al. Epidemiology of antineutrophil cytoplasmic antibody-associated vasculitis. Rheumatic Disease Clinics of North America, v. 41, n. 1, p. 1–19, 2015.
YANG, H. et al. Pulmonary involvement in ANCA-associated vasculitis: Pathogenesis, clinical features and treatment. Journal of Thoracic Disease, v. 9, n. 11, 2017.
ZHANG, Y. et al. (2024). Pulmonary involvement in microscopic polyangiitis: A retrospective analysis of 273 patients. BMC Pulmonary Medicine, v. 24, n. 1, p. 1-10, 2024.
1Residente em Clínica Médica
Endereço: Maringá – Paraná, Brasil
E-mail: nemenahhh@gmail.com
2Especialista em Clínica Médica
Coordenador da Residência de Clinica Medica do Hospital Memorial Uningá
Coordenador do Núcleo Interno da Regulação dos Leitos do Hospital Memorial Uningá
Endereço: Maringá – Paraná, Brasil
E-mail: prof.jerdalmorsoletto@uninga.edu.br
3Especialista em Hematologia
Diretor do Comitê Transfusional e de óbitos do Hospital Memorial da Uningá
Preceptor do Ambulatório de Hematologia Unidade Memorial II
Endereço: Maringá – Paraná, Brasil
E-mail: francismarprestesleal@gmail.com
4Especialista em Clínica Médica
Endereço: Maringá – Paraná, Brasil
E-mail: dr.gabrielnoliveira@gmail.com
5Coordenador do Curso de Medicina Uningá
Diretor Médico das Unidades Hospitalar Memorial I e Ambulatorial II
Endereço: Maringá – Paraná, Brasil
E-mail: zeliofedattojr@yahoo.com.br