APPROACH TO DUCHENNE MUSCULAR DYSTROPHY IN PRIMARY CARE – CASE REPORT

REGISTRO DOI: 10.69849/revistaft/fa10202501291021
Marina Queiroz Tobias Costa1,
Orientadora: Bruna Evellyn de Lima Alves2
RESUMO
Objetivo: Descrever um caso de Distrofia Muscular de Duchenne e discutir o quadro clínico e a abordagem diagnóstica na Atenção Primária à Saúde. Métodos: Revisão de prontuário e de literatura. Detalhamento de Caso: Paciente do sexo masculino, 12 ano, filho de pais não consanguíneos sem histórico familiar conhecido para a doença. Há 4 anos apresentando quadro de quedas frequentes associado a redução da força em membros inferiores. Devido ao quadro, procurou acompanhamento na UBS da região. Ao exame físico, apresentava hipertrofia das panturrilhas, sinal de Gowers, marcha alterada, dificuldade de subir degraus e andar nas pontas dos pés. Exames laboratoriais demonstraram creatinofosfoquinase 6052 U/I, TGO 73,3 e TGP 68,3. No teste genético apresentou deleção do éxon 50 no gene da distrofina, caracterizando a doença. Conclusão: É importante o reconhecimento precoce das manifestações clínicas por profissionais de saúde das unidades da atenção primária em saúde, pois através do acompanhamento e identificação o quanto antes dos sinais e sintomas, há maior chance de retardar a progressão da doença ao instituir brevemente o tratamento com a equipe multidisciplinar objetivando melhor qualidade de vida e maior expectativa de vida.
Palavras-chave: distrofia muscular de duchenne, atenção primária à saúde, diagnóstico.
INTRODUÇÃO
A distrofia muscular de Duchenne é uma rara e grave doença com acometimento progressivo com evolução para atrofia muscular. É uma distrofinopatia de caráter genético associado a distúrbio recessivo ligado ao cromossomo X, com estimativa de 1 em 6.000 nascidos vivos do sexo masculino. Ocasionada por mutações na codificação da distrofina, impedindo a produção da isoforma muscular e causando o truncamento prematuro da tradução de proteínas, levando à distrofina não funcional e instável. (DUAN et al., 2021 e ARORA, H., 2019)
Em relação as manifestações clínicas, é importante suspeitar de crianças do sexo masculino com idade entre 2 a 4 anos com atraso nos marcos motores, fraqueza muscular, panturrilhas hipertróficas e o sinal de Gowers. Este sinal corresponde ao paciente necessitar do uso das mãos para tentar se levantar do chão para compensar a fraqueza muscular da região superior da perna e quadril. Além do acometimento motor, pode ocorrer comprometimento cognitivo com atraso na fala também. (BUSHBY, K. et al.,2010 e FOX, H. et al.,2020)
Há um padrão de fraqueza muscular proximal progressivo, iniciando pelos membros inferiores. É importante estar atento aos marcos motores, como andar, dificuldade de pular, correr, subir degraus, levantar-se do chão. Pode haver também relato de mialgia, marcha anormal, andar nas pontas dos pés e quedas frequentes. (ARAUJO, A. P. Q. C. et al., 2017 e PRUFER, A. et al.,2023)
FOX, H. et al. (2020) citaram para auxiliar no diagnóstico, um mnemônico MUSCLE, caracterizado por:
Motor milestone (marco motor atrasado)
Incapaz de andar aos 18 meses
Incapaz de pular aos 2 anos e meio
Incapaz de correr aos 3 anos
Unusual gait (marcha incomum)
Caminhar nas pontas dos pés
Quedas frequentes
Dificuldade de subir degraus
Speech delay (atraso na fala)
Nenhuma palavra falada nos primeiros 18 meses
Incapaz de falar frases aos 3 anos
Acompanhamento em serviços de fala e linguagem
Creatine kinase asap (dosagem creatinofosfoquinase)
Leads to (leva a)
Early diagnosis of DMD (diagnóstico precoce de DMD)
Se apresentar mais de 3 sintomas de 2 ou mais categorias, recomenda-se a dosagem de creatinofosfoquinase. Além disso, é importante observar os sinais de alerta: hipertrofia das panturrilhas, episódio de mioglobinúria e dor muscular ou cãibras. (FOX, H. et al.,2020)
Idealmente, recomenda-se o diagnóstico o mais precoce possível em torno de 2 a 3 anos. Se suspeita de DMD (distrofia muscular de Duchenne), deve ser solicitado alguns exames. Sobre as alterações laboratoriais que possam estar relacionadas estão a dosagem de cpk (creatinofosfoquinase), tgo (transaminase glutâmico-oxalacética) e tgp (transaminase glutâmico-pirúvica) elevadas, devido ao dano muscular contínuo e à alta quantidade dessas enzimas no músculo esquelético. (FALZARANO, M. et al.,2015 e MERCURI, E.; et al.,2018)
Diagnóstico é confirmado através de testes genéticos. Cerca de 70% dos casos são ocasionados por deleção ou duplicação. É importante também investigação do acometimento dos outros sistemas. Realização de espirometria faz-se necessário pois há risco de complicações respiratórias repentinas, tais como insuficiência respiratória. (OSORIO, A. et al.,2018)
Na abordagem terapêutica, ainda não há cura para a distrofia muscular de Duchenne. Entretanto, há medidas realizadas a partir da multidisciplinaridade visando melhora dos sintomas, bem como a qualidade de vida. Além da equipe médica, é necessário que os enfermeiros, fisioterapeutas, nutricionistas, psicólogos, terapeutas ocupacionais e assistente social auxiliem e participem da terapêutica com o objetivo de oferecer cuidado integral ao paciente. (ARORA, H.,2019)
Nessa perspectiva, ressalta-se a importância da integração da rede de cuidado, promovendo acesso, equidade, qualidade, atenção contínua e longitudinal. Tais características são importantes na coordenação do cuidado. É a articulação de ações de saúde, sincronizada, atenção integrada nos diversos níveis de forma vertical, mas mantendo assistência na APS e serviços de saúde na dimensão horizontal. (RIBEIRO; CAVALCANTI, 2020)
Esse estudo tem como objetivo descrever a investigação e a abordagem realizada em uma criança Unidade Básica de Saúde (UBS) com suspeita e posteriormente confirmação do diagnóstico de DMD. Assim como detalhar seu acompanhamento com a atenção primária à saúde e sua articulação e condução do quadro juntamente com a atenção secundária, especialistas focais, tais como a neuropediatria, ortopedia, endocrinologia e a equipe multidisciplinar. Dessa forma, ressalta-se a importância da integralidade e da longitudinalidade na coordenação do cuidado.
DETALHAMENTO DO CASO:
O caso em questão é um estudo aprovado pelo Comitê de Ética em Pesquisa (CEP) sob parecer número 7.271.198 e CAAE 83889624.1.0000.5553, após leitura e assinatura pelo responsável do paciente do Termo de Consentimento Livre e Esclarecido (TCLE), bem como Termo de Assentimento Livre e Esclarecido (TALE). Criança, 12 anos, masculino.
Comparece à UBS com queixa de redução da força em membros inferiores com quedas frequentes há 4 anos. Sobre antecedentes, nasceu de parto normal, apgar 9/10, idade gestacional de 40 semanas, peso 2955 gramas, sorologias sífilis e HIV negativos, realizado duas consultas de pré- natal ainda durante o primeiro trimestre. Sem demais informações. Pais usuários de substâncias ilícitas há cerca de 10 anos, familiares não sabem informar se ocorreu exposição durante a gestação e as substâncias usadas. Histórico da criança ter permanecido em abrigo durante oito anos. Não há informações sobre amamentação, marcos do desenvolvimento neuropsicomotor, introdução alimentar e suplementação durante a primeira infância. Vacinação atualizada segundo o Programa Nacional de Imunização.
Possui quatros irmãos, sendo três meninas e um menino que não apresentam sintomas e sinais semelhantes ao quadro do paciente, hígidos. Durante a anamnese, está acompanhado do seu representante legal que informa perda progressiva da forma em membros inferiores com atrofia em coxas bilateral, associado a quedas frequentes, dificuldade de subir degraus e aumento da região da panturrilha. Nega outras queixas associadas.
Avaliado em conjunto com a fisioterapeuta da equipe multiprofissional da Atenção Primária à Saúde (e-Multi) com resultado de avaliação motora: sinal de Gowers positivo – piora do quadro há 3 anos, aumento da circunferência da panturrilha, fraqueza muscular global, alteração da marcha com instabilidade de tronco, pouca dissociação de cinturas, não realiza toque do calcanhar, apresenta fraqueza global alterando todas as fases da marcha. Dificuldade nas AVD’s (atividades da vida diária). Medida de função motora: realiza completamente o movimento solicitado de modo imperfeito, déficit de equilíbrio.
Ao exame físico apresenta bom estado geral, vigil, consciente e orientado em tempo e espaço, afebril, anictérico, acianótico. Sinais vitais: pressão arterial 100×60 mmHg, frequência cardíaca 67 bpm, frequência respiratória 18 irpm, saturação de oxigênio 98%, temperatura 36ºC. Aparelho respiratório: sem alterações. Aparelho cardiovascular: sem alterações. Abdome: sem alterações. Membros inferiores: hipertrofia de panturrilhas, atrofia da região proximal do membro (coxa) com redução da força em ambos os membros inferiores. Neurológico: força grau 4- proximal em membros superiores, grau 4+ distal em membros superiores, grau 3 proximal em membros inferiores, grau 4- na musculatura anterior de membros inferiores e 4+ em musculatura posterior. Marcha anserina, com báscula de quadril e hiperlordose. Sinal de Gowers positivo.
Segue abaixo imagens da hipertrofia das panturrilhas, segundo figura 1 e 2:
Figura 1 – Imagem que evidencia aumento da panturrilha (anterior):
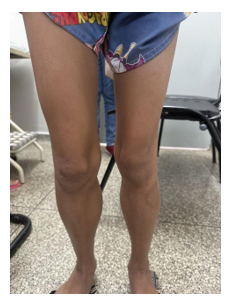
Fonte: acervo pessoal
Figura 2 – Imagem que evidencia aumento da panturrilha (posterior):
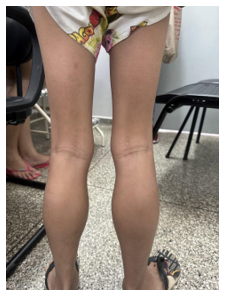
Fonte: acervo pessoal
Em relação aos exames complementares laboratoriais com valores:
Tabela 1 – Valores dos exames laboratoriais
Exames laboratoriais Valor CPK 6052 TGP 68,3 TGO 73,3 Cálcio 10,3 CKMB 176 Creatinina 0,32 DHL 370 Fosfatase alcalina 113 Fósforo 5,7 Glicemia 77 Ureia 21 Hemoglobina 13,4 Leucócitos 7300 Plaquetas 378000 TSH 3,84
Desidrogenase láctica (DHL); hormônio tireoestimulante (TSH)
Foram solicitados ressonância cardíaca, ecocardiograma, eletrocardiograma, espirometria e ressonância magnética cerebral pelo sistema nacional de regulação com período entre a solicitação e execução em torno de 2 semanas. Os exames foram realizados e sem alterações. Além da solicitação pelo sistema de regulação dos exames, foi demandado também o encaminhamento à neurologia pediátrica, ortopedia e endocrinologia com a primeira consulta realizada pela neurologia pediátrica em 2 semanas.
Iniciado albendazol 400 mg durante 3 dias para início posterior de corticoterapia com prednisolona 3 mg/ml 6,5 ml ao dia e enalapril 5 mg 1 comprimido pela manhã de forma contínua. Recomendado vacinação para influenza, covid 19 e pneumocócica 23.
Realizado teste genético com o painel DNAmplo com deleção do éxon 50, sendo identificada hemizigose no gene DMD (gene da distrofina). Em seguida, foi solicitado a confecção da cadeira de rodas adaptada e órteses para os membros inferiores. Mantém seguimento conjunto com a atenção primária à saúde e os especialistas, neurologia pediátrica, ortopedista, endocrinologista e equipe multidisciplinar com a terapeuta ocupacional, fisioterapia, psicologia, fonoaudiologia e assistente social.
DISCUSSÃO
No presente caso, a criança nasceu bem e não houve relatos sobre anormalidade no desenvolvimento neuropsicomotor devido a limitação de registros na caderneta da criança por ficar cerca de 8 anos em abrigo, sem acompanhamento dos familiares. Aos 8 anos de idade, após estar sob a guarda judicial da avó, percebeu-se quedas frequentes e instabilidade postural, com dificuldade de subir degraus e andar nas pontas dos pés.
As manifestações clínicas do paciente são compatíveis com os sintomas achados na DMD na literatura. Músculos da cintura pélvica sendo acometidos ocasionando o desequilíbrio, instabilidade e frequentes quedas. Surgimento de contratura nos tendões de Aquiles levando a andar nas pontas dos pés. Os achados do exame físico também revelam a hipótese diagnóstica devido a pseudohipertrofia dos músculos gastrocnemios, sinal de Gowers e alteração de marcha. (BIRNKRANT, D. J. et al.,2019 e ANNEXSTAD, E. J.; et al.,2014)
Ao solicitar os exames laboratoriais, foram obtidos resultados que corroboram para o diagnóstico precoce. Elevação dos valores de CPK, TGO, TGP e LDH. Isso ocorre devido a liberação a partir dos músculos afetados. Realizado também o teste genético onde apresentou alterações, tais como a deleção do éxon 50, sendo identificada hemizigose no gene DMD (gene da distrofina), de acordo com achados esperados na literatura. (YIU, E. M.; KORNBERG, A. J.,2015)
Devido a ausência de distrofina no músculo cardíaco, é necessário a avaliação cardiológica através do eletrocardiograma e ecocardiograma ou ressonância magnética cardíaca anual para pesquisar o desenvolvimento de cardiomiopatia dilatada progressiva, ocasionando insuficiência cardíaca congestiva, insuficiência cardíaca, anormalidades de condução, arritmia ventricular ou supraventricular e risco de morte súbita precoce. Exames executados sem alterações. Foi solicitado também espirometria para avaliação da função respiratória pois pode haver associação e evolução para insuficiência respiratória. Exame sem anormalidades. (BIRNKRANT, D. J. et al.,2018; BUDDHE, S. et al.,2018 e MCNALLY, E. M. et al.,2015)
Algumas referências relatam correlação do comprometimento cognitivo e dificuldades de aprendizagem e problemas comportamentais com volume reduzido de substância cinzenta, anormalidades de substância branca e perfusão cerebral reduzida vistos pela ressonância magnética cerebral. Sugere-se que no cérebro, a distrofina é expressa nos neurônios GABAérgicos inibitórios pós-sinápticos na amígdala, hipocampo e células de Purkinje cerebelares, correlacionando o comprometimento cognitivo com mutações que afetam as isoformas mais curtas da distrofina. Foi realizado o exame, sem alterações. (BANIHANI, R. et al., 2015 e CHIEFFO, D. et al.,2015)
A DMD geralmente é diagnosticada entre 3 e 7 anos, período em que os responsáveis observam um possível atraso no desenvolvimento. No atual caso, pelo fato da criança estar em um abrigo por anos e nos últimos 4 anos observarem alterações motoras e de difícil reconhecimento por outros profissionais de saúde que passou, possibilitou um diagnóstico tardio. (FALZARANO, M. et al., 2015; FOX, H. et al.,2020 e RYDER, S. et al., 2017)
As mais recentes diretrizes recomendam o uso de corticoides, como a prednisona visando melhorar a força, função motora, função pulmonar, retardar a perda da deambulação com média de 2 anos e início da cardiomiopatia, bem como reduz a necessidade de abordagem cirúrgica para escoliose. Não se sabe o mecanismo exato que os glicocorticosteroides atrasam a progressão da doença, entretanto há hipóteses de que os glicocorticosteroides diminuem a inflamação e aumentam a massa muscular total e a força através da estimulação de fatores de crescimento semelhantes à insulina, diminuição da produção de citocinas, diminuição da reação dos linfócitos, aumento da proliferação de mioblastos e regulação positiva de moléculas sinérgicas. Foi iniciado corticoterapia com prednisolona 3 mg/ml 6,5 ml ao dia. Segundo estudos, corticoide auxilia a melhorar a força, função motora, função pulmonar, retardar a perda da deambulação e o início da cardiomiopatia. (KORINTHENBERG, R., 2018; MATTHEWS, E. et al.,2016 e MCDONALD, C. M. et al., 2018)
Se ocorrer sintomas de insuficiência cardíaca e arritmias, poderá ser utilizados os inibidores da enzima conversora de angiotensina (IECA) e betabloqueadores. A introdução precoce de inibidores da ECA como tratamento cardioprotetor é sugerido aos pacientes em torno dos 10 anos de idade, pois o tratamento profilático é recomendado para retardar o início dos sintomas cardíacos. Foi iniciado também o enalapril 5 mg 1 comprimido pela manhã de forma contínua, pois possui função cardioprotetora. (REINIG, A. M.; et al.,2017 e SUN, C. et al.,2020)
Além disso, a avaliação ortopédica também é indispensável visando a prevenção de contrações dos tendões de Aquiles, flexores do joelho e do quadril e bandas iliotibiais através do suporte da fisioterapia e do uso de órteses para preservar a estabilidade e a capacidade de deambular. É importante mencionar a supervisão da saúde óssea devido ao acometimento pelo tratamento crônico com o uso dos corticoides como efeito colateral. (BIRNKRANT, D. J. et al.,2018)
Devido a evolução no desenvolvimento da terapêutica e aprimoramento da abordagem, pacientes com distrofia muscular de Duchenne possuem aumento da expectativa de vida média, comparado a anos atrás. Anteriormente, expectativa de vida média era de 25 anos para aqueles nascidos antes de 1970 e 40 anos para aqueles nascidos depois de 1970. Reflexo dos resultados obtidos por meio das melhorias relacionadas ao tratamento e acompanhamento dos pacientes. (KIENY P, et al.,2013 e SAITO, T. et al.,2017)
Por ser uma doença genética ligada ao cromossomo X, vale ressaltar o papel do médico da atenção primária em acompanhar e orientar sobre planejamento familiar e aconselhamento reprodutivo das mulheres portadoras da mutação do gene ao identificá-las. Além disso, uma porcentagem dessas portadoras podem desenvolver sintomas musculares como mialgia, cãibras, fraqueza, problemas de aprendizagem ou comportamentais. (FOX, H. et al.,2020)
Avaliação integrada com a endocrinológica torna-se essencial devido ao acompanhamento conjunto dos possíveis efeitos colaterais do uso crônico da corticoterapia, como o risco de osteoporose, puberdade tardia, obesidade, diabetes mellitus, juntamente com a nutrição. Ressaltando a recomendação de suplementação de vitamina D e ingestão adequada de cálcio. (BIRNKRANT, D. J. et al.,2018)
Estudos informam comprometimento a longo prazo do trato gastrointestinal também por acometimento dos músculos lisos viscerais. Com isso, pacientes com DMD podem desenvolver lentificação do esvaziamento gástrico e paresia intestinal com doença do refluxo gastroesofágico e constipação. Entretanto, esse quadro torna-se mais comum em adultos. (BIRNKRANT, D. J. et al.,2018)
Paciente segue sendo acompanhado pela equipe multidisciplinar. Além do médico da família e comunidade, neurologista pediátrico, endocrinologista e ortopedista. Também está sendo acolhido pela fisioterapeuta, terapeuta ocupacional, psicóloga e fonoaudióloga. Tendo como o objetivo o prolongamento e melhora na qualidade de vida.
Percebe-se também o papel da atenção primária à saúde em oferecer suporte ao garantir a vacinação adequada como a vacina pneumocócica e a vacina anual para influenza, monitorar os efeitos colaterais do uso dos corticoides , monitorar peso, considerar gastroproteção aos pacientes que possuem sintomas em uso crônico de corticoides, acompanhamento conjunto com os especialistas, monitorar os sinais de alerta de deterioração clínica e proporcionar acesso a cuidados psicossociais para o paciente e seus familiares. (FOX, H. et al.,2020)
CONSIDERAÇÕES FINAIS
Nota-se portanto, a importância do reconhecimento precoce das manifestações clínicas por profissionais de saúde das unidades da atenção primária em saúde também por ser a via de primeiro acesso do usuário. Nota-se que através do acompanhamento e identificação o quanto antes dos sinais e sintomas, há maior chance de retardar a progressão da doença ao instituir brevemente o tratamento com a equipe multidisciplinar objetivando melhor qualidade de vida e maior expectativa de vida.
REFERÊNCIAS
- ANNEXSTAD, E. J.; et al. Duchennes muskeldystrofi. Tidsskrift for Den norske legeforening, v. 134, n. 14, p. 1361–1364, 2014.
- ARAUJO, A. P. Q. C. et al. Brazilian consensus on Duchenne muscular dystrophy. Part 1: diagnosis, steroid therapy and perspectives. Arquivos de Neuro-Psiquiatria, v. 75, n. 8, p. 104–113, ago. 2017.
- ARORA, H. Duchenne muscular dystrophy: Still an incurable disease. Neurology India, v. 67, n. 3, p. 717, maio 2019.
- BANIHANI, R. et al. Cognitive and Neurobehavioral Profile in Boys With Duchenne Muscular Dystrophy. Journal of Child Neurology, v. 30, n. 11, p. 1472–1482, 6 fev. 2015.
- BIRNKRANT, D. J. et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. The Lancet Neurology, v. 17, n. 3, p. 251–267, mar. 2018.
- BIRNKRANT, D. J. et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. The Lancet Neurology, v. 17, n. 4, p. 347–361, abr. 2018.
- BUDDHE, S. et al. Cardiac Management of the Patient With Duchenne Muscular Dystrophy. Pediatrics, v. 142, n. Supplement 2, p. S72–S81, out. 2018.
- BUSHBY, K. et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. The Lancet Neurology, v. 9, n. 1, p. 77–93, jan. 2010.
- CHIEFFO, D. et al. Early Neurodevelopmental Findings Predict School Age Cognitive Abilities in Duchenne Muscular Dystrophy: A Longitudinal Study. PLOS ONE, v. 10, n. 8, p. e0133214, 14 ago. 2015.
- DUAN, D. et al. Duchenne Muscular Dystrophy. Nature Reviews Disease Primers, v. 7, n. 1, p. 1–19, 18 fev. 2021.
- FALZARANO, M. et al. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules, v. 20, n. 10, p. 18168–18184, 7 out. 2015.
- FOX, H. et al. Duchenne muscular dystrophy. BMJ, p. l7012, 23 jan. 2020.
- KIENY P, et al. Evolution of life expectancy of patients with Duchenne muscular dystrophy at AFM Yolaine de Kepper centre between 1981 and 2011. Annals of Physical and Rehabilitation Medicine, v. 56, n. 6, p. 443–454, 1 set. 2013.
- KORINTHENBERG, R. A new era in the management of Duchenne muscular dystrophy. Developmental Medicine & Child Neurology, v. 61, n. 3, 16 dez. 2018.
- MATTHEWS, E. et al. Corticosteroids for the treatment of Duchenne muscular dystrophy. The Cochrane database of systematic reviews, v. 5, n. 5, p. CD003725, 5 maio 2016.
- MCDONALD, C. M. et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. The Lancet, v. 391, n. 10119, p. 451–461, fev. 2018.
- MCNALLY, E. M. et al. Contemporary Cardiac Issues in Duchenne Muscular Dystrophy. Circulation, v. 131, n. 18, p. 1590–1598, 5 maio 2015.
- MERCURI, E.; et al. Muscular Dystrophies. The Lancet, v. 394, n. 10213, p. 2025–2038, nov. 2019.
- OSORIO, A. et al. Consenso para el diagnóstico, tratamiento y seguimiento del paciente con distrofia muscular de Duchenne. Neurología, v. 34, n. 7, mar. 2018.
- PRUFER, A. et al. Update of the Brazilian consensus recommendations on Duchenne muscular dystrophy. v. 81, n. 01, p. 081–094, 1 jan. 2023.
- REINIG, A. M.; et al. Advances in the Treatment of Duchenne Muscular Dystrophy: New and Emerging Pharmacotherapies. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, v. 37, n. 4, p. 492–499, 10 mar. 2017.
- RIBEIRO, S. P.; CAVALCANTI, M. DE L. T. Atenção Primária e Coordenação do Cuidado: dispositivo para ampliação do acesso e a melhoria da qualidade. Ciência & Saúde Coletiva, v. 25, n. 5, p. 1799–1808, maio 2020.
- RYDER, S. et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet Journal of Rare Diseases, v. 12, n. 1, 26 abr. 2017.
- SAITO, T. et al. Study of Duchenne muscular dystrophy long-term survivors aged 40 years and older living in specialized institutions in Japan. Neuromuscular Disorders, v. 27, n. 2, p. 107–114, 1 fev. 2017.
- SUN, C. et al. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes, v. 11, n. 8, 23 jul. 2020.
- YIU, E. M.; KORNBERG, A. J. Duchenne muscular dystrophy. Journal of Paediatrics and Child Health, v. 51, n. 8, p. 759–764, 9 mar. 2015.
1Residente do segundo ano do programa de residência médica em Medicina da Família e Comunidade da ESCS-FEPECS
2Orientadora e preceptora do programa de residência médica em Medicina da Família e Comunidade da ESCS-FEPECS