REGISTRO DOI: 10.69849/revistaft/pa10202412161436
Júnia Marise Ramos1
Juliana Lilis da Silva2
Natália de Fátima Gonçalves Amâncio2
Juliana Ribeiro Gouveia Reis2
Resumo:
Introdução: A fibrose cística (FC) é uma doença genética grave, causada por mutações no gene CFTR. No Brasil, ocorre em 1 a cada 10 mil nascimentos. O diagnóstico é feito por testes como o do pezinho, do suor e sequenciamento genético. O tratamento é multidisciplinar, com ênfase em fisioterapia respiratória, nutrição e medicamentos, as complicações pulmonares são as principais causas de morbidade e mortalidade. Objetivo: Avaliar os benefícios das terapias respiratórias na redução das complicações pulmonares e melhoria da qualidade de vida. Metodologia: Realizou-se uma revisão integrativa sobre terapias respiratórias na FC, focando na qualidade de vida e complicações pulmonares. Foram analisados artigos de 2020 a 2024, com critérios específicos de inclusão e exclusão. Resultados e Discussão: Após o levantamento das publicações, 27 artigos foram listados e 7 excluídos. A FC é uma doença grave que pode levar à morte precoce, principalmente por infecções pulmonares. Exige tratamento contínuo e multidisciplinar, com foco em estabilização clínica e bem-estar psicológico. As terapias respiratórias, como drenagem postural, pressão expiratória positiva, compressão torácica de alta frequência, tapotagem, vibração, vibrocompressão, técnica de expiração forçada, huffing, oscilação oral de alta frequência, drenagem autógena, ciclo ativo de respiração, expiração lenta e prolongada, aumento do fluxo expiratório, hiperinsuflação manual, aspiração traqueal, expiração lenta total com a glote aberta em decúbito lateral, ajudam a melhorar a função pulmonar e a reduzir exacerbações. Conclusão: As terapias respiratórias, quando aplicadas por profissionais capacitados, são eficazes para a melhora da função pulmonar, redução de exacerbações e melhora da qualidade de vida.
Palavras-chaves: Fibrose Cística. Terapia respiratória. Qualidade de vida.
1 INTRODUÇÃO
A fibrose cística (FC) é uma doença genética grave, progressiva, determinada por um padrão de herança autossômico recessivo, com acometimento multissistêmico e impacto significativo na qualidade e na expectativa de vida dos pacientes. Causada por mutações no gene CFTR, a fibrose cística resulta em disfunção na proteína CFTR, afetando diversas glândulas exócrinas e tecidos (SANTOS, et al 2024).
Nos Estados Unidos ocorre em cerca de 1/3.300 nascimentos de brancos, 1/15.300 nascimentos de negros e 1/32.000 nascimentos de americanos de origem asiática. Estima-se que cerca de 40.000 pessoas nos EUA vivam com a condição, e cerca de 100.000 em todo o mundo (MANUAL MSD, 2023). No Brasil é uma das doenças raras mais comuns, atingindo 1 a cada 10 mil nascidos vivos no país. Graças aos avanços científicos e a melhoria da assistência aos pacientes, a sobrevida vem aumentando a cada ano. Dados de registros internacionais apontam uma sobrevida média atual de cerca de 40 anos (MINISTÉRIO DA SAÚDE, 2022).
A fisiopatologia da fibrose cística é causada por variantes patogênicas no gene CFTR, que codifica uma proteína responsável pelo transporte de cloreto e bicarbonato nas células epiteliais. Defeitos na função dessa proteína resultam em diversas manifestações clínicas, incluindo insuficiência pancreática, má absorção de nutrientes, doença pulmonar crônica e aumento do risco de desidratação, devido à elevada concentração de eletrólitos no suor. As alterações no sistema respiratório são as principais responsáveis pela morbidade e mortalidade associadas à doença ZAMBERLAN, 2023).
O cenário da fibrose cística, tanto no Brasil quanto no mundo, tem passado por transformações devido à introdução de novas tecnologias voltadas para o diagnóstico e o tratamento da doença (DUTRA, et al 2020).
O diagnóstico da fibrose cística é realizado primeiramente através do teste do pezinho, conhecido como triagem neonatal, que detecta níveis elevados de imunorreatividade de tripsina imunorreativa (IRT), Em seguida realiza-se o teste do suor onde mede-se a concentração de cloreto de sódio no suor, pacientes com fibrose cística geralmente apresentam valores mais altos que indivíduos saudáveis. Por último realiza-se o sequenciamento genético para identificar as mutações do gene CFTR responsável pela fibrose cística (SILVA, 2024).
O tratamento é realizado de forma multidisciplinar e consiste em suporte nutricional, fisioterapia respiratória e uso de medicamentos. A adesão ao tratamento representa um desafio em razão da complexidade e do número de intervenções exigidas, o que requer um suporte contínuo e uma abordagem multidisciplinar cujo objetivos são retardar a progressão da doença e minimizar os impactos das complicações (RUIZ; LEAL, 2023).
As complicações pulmonares são as principais causas de morbimortalidade dos pacientes com fibrose cística. A obstrução das vias áreas causada pela produção excessiva de secreção propicia quadros de inflamação crônica e infecções recorrentes, afetando a força muscular respiratória, que associada ao comprometimento da capacidade ventilatória pode resultar em fadiga, intolerância ao exercício e dificuldades nas atividades de vida diária, iniciando um ciclo vicioso em que a piora da dispneia está associada à diminuição da atividade física, comprometendo a qualidade de vida dessa população (MUCHA, et al 2020).
Apesar dos avanços científicos e terapêuticos que têm proporcionado melhorias significativas na qualidade de vida, ainda existem obstáculos a serem superados, como o acesso a tratamentos especializados e a individualização dos cuidados. As perspectivas futuras são promissoras, com o desenvolvimento de terapias inovadoras e a busca por abordagens personalizadas (RUIZ; LEAL, 2023).
Visto que a fibrose cística é uma condição complexa que exige estratégias abrangentes e inovadoras para aumentar a qualidade de vida e prolongar a sobrevida dos pacientes que enfrentam essa doença genética grave, justifica-se a realização deste estudo buscando apresentar as terapias clínicas respiratórias para melhor prognóstico da doença e qualidade de vida.
Desta forma o presente estudo tem por objetivo revisar na literatura estudos recentes que evidenciam os benefícios das terapias clínicas respiratórias no manejo da fibrose cística como instrumento para redução de complicações pulmonares e melhora da qualidade de vida desses pacientes.
2 METODOLOGIA
O presente estudo consiste de uma revisão exploratória integrativa de literatura. A revisão integrativa foi realizada em seis etapas: 1) identificação do tema e seleção da questão norteadora da pesquisa; 2) estabelecimento de critérios para inclusão e exclusão de estudos e busca na literatura; 3) definição das informações a serem extraídas dos estudos selecionados; 4) categorização dos estudos; 5) avaliação dos estudos incluídos na revisão integrativa e interpretação e 6) apresentação da revisão.
Na etapa inicial, para definição da questão de pesquisa utilizou-se da estratégia PICO (Acrômio para Patient, Intervention, Comparation e Outcome). Assim, definiu-se a seguinte questão central que orientou o estudo: “Quais são as terapias clínicas respiratórias disponibilizadas no tratamento da Fibrose Cística, para melhoria da qualidade de vida e redução de complicações pulmonares?” Nela, observa-se o P: Pacientes com fibrose cística; I: Terapias clínicas respiratórias; C: Não se aplica; O: Melhora da qualidade/expectativa de vida, redução de complicações pulmonares.
Para responder a esta pergunta, foi realizada a busca de artigos envolvendo o desfecho pretendido utilizando as terminologias cadastradas nos Descritores em Ciências da Saúde (DeCs) criados pela Biblioteca Virtual em Saúde desenvolvido a partir do Medical Subject Headings da U.S. National Library of Medcine, que permite o uso da terminologia comum em português, inglês e espanhol. Os descritores utilizados foram: fibrose cística, terapias clínicas, complicações pulmonares, qualidade de vida, expectativa de vida. Para o cruzamento das palavras chaves utilizou-se os operadores booleanos “and”, “or” “not”.
Realizou-se um levantamento bibliográfico por meio de buscas eletrônicas nas seguintes bases de dados: Google Scholar; Biblioteca Virtual de Saúde (BVS), Scientif Eletronic Library Online (SciELO), National Library of Medicine (PubMed), EbscoHost.
A busca foi realizada no período de agosto a outubro de 2024. Como critérios de inclusão, limitou-se a artigos escritos em português, inglês e espanhol publicados nos últimos cinco anos (2020 a 2024), que abordassem o tema pesquisado e que estivem disponíveis eletronicamente em seu formato integral, foram excluídos os artigos em que o título e resumo não estivessem relacionados ao tema de pesquisa e pesquisas que não tiverem metodologia bem clara.
Posteriormente a seleção dos artigos, foi realizado um fichamento das obras selecionadas afim de selecionar a coleta e análise dos dados. Os dados coletados foram disponibilizados em um quadro, possibilitando ao leitor a avaliação da aplicabilidade da revisão integrativa elaborada, de forma a atingir o objetivo desse método.
3 RESULTADOS
Após a etapa de levantamento das publicações, foram listados 27 artigos, dos quais foram realizados a leitura do título e resumo das publicações considerando o critério de inclusão e exclusão definidos. Em seguida, realizou a leitura na íntegra das publicações, atentando-se novamente aos critérios de inclusão e exclusão, sendo que 7 artigos não foram utilizados devido aos critérios de exclusão. Foram selecionados 20 artigos para análise final e construção da revisão.
A Figura 1 demonstra o processo de seleção dos artigos por meio das palavras-chaves de busca e da aplicação dos critérios de inclusão e exclusão citados na metodologia. O fluxograma leva em consideração os critérios elencados pela estratégia PRISMA (Page et al., 2021).
Figura 1 – Fluxograma da busca e inclusão dos artigos
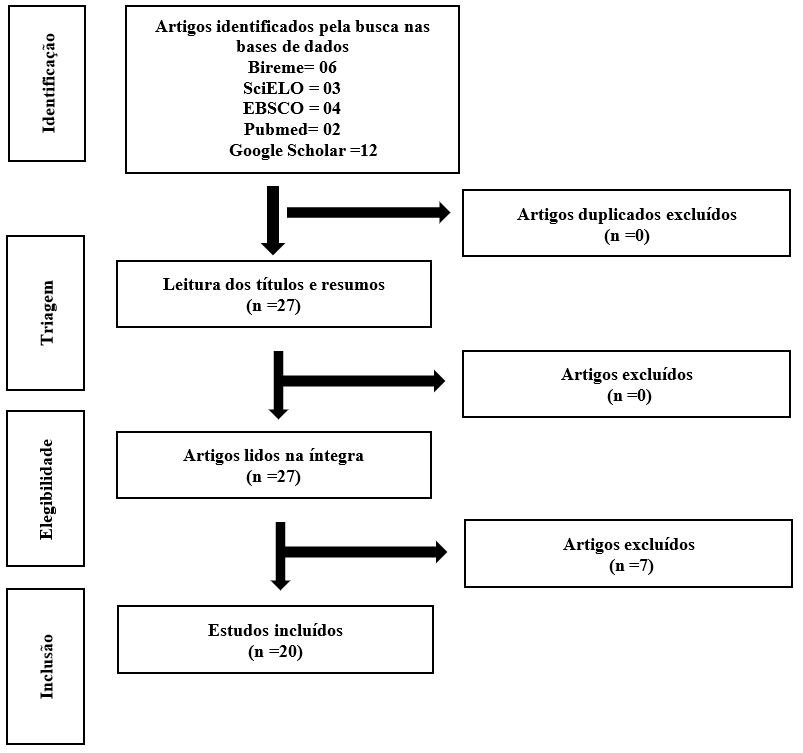
Fonte: Fonte: Adaptado do Preferred Reporting Items for Systematic review and Meta-Analyses (PRISMA). Page et al., (2021).
A Tabela 1 sintetiza os principais artigos que foram utilizados na presente revisão de literatura, contendo informações relevantes, como os autores do estudo, o ano de publicação, o título e os achados relevantes.
Tabela 1 – Terapias clínicas oferecidas para pacientes com Fibrose Cística encontradas nas publicações no período de 2020 a 2024.
AUTOR, ANO TÍTULO MÉTODO ACHADOS PRINCIPAIS GRYSZTAR et al., 2024 Polish Cystic Fibrosis Patients’ Health-Related Quality of Life and Its Influencing Factors: A Cross-Sectional, Single-Centre Study Aplicação do questionário Cystic Fibrosis Questionnaire-Revised (CFQ-R). Exacerbações da doença reduziram a QVRS. SANTOS et al.,2024 Perspectivas futuras em terapias moduladoras para Fibrose Cística: Avanços e desafios Terapia com moduladores do CFTR Avanço no tratamento e melhora da qualidade de vida. REIS, NEVES, 2024 Terapias de higiene brônquica durante exacerbações de pacientes com fibrose cística: uma revisão de literatura Ciclo ativo das técnicas respiratórias (CATR), vibrocompressão, tapotagem, drenagem postural, técnica de expiração forçada (TEF), exercícios de expansão torácica (EET), tosse e/ou huffing. Todas as técnicas para higiene brônquica são benéficas DAINAVAS et al., 2024 The Impact of Cost of Living on the Quality of Life of Cystic Fibrosis Patients: A Study in Greece Questionários Short Form Questionnaire-36 (SF-36) e versão grega do Cystic Fibrosis Quality of Life (CFQoL). Menores custos gerais devido a doença, melhor qualidade de vida. LANFRANCHI et al., 2024 A case of severe pulmonary exacerbation in a young patient with cystic fibrosis in the era of CFTR modulators Terapias com moduladores do CFTR. Pouca compreensão das exacerbações respiratórias. UPADHYAY et al., 2024 Current and future therapeutic approaches of CFTR and airway dysbiosis in an era of personalized medicine Terapias moduladoras do CFTR e papel do microbioma das vias aéreas. Importância de entender as causas celulares e moleculares da FC para melhoria da qualidade de vida. SILVA, SOUTO, 2024 Fibrose Cística – Principais características clínicas e métodos de diagnóstico Teste do suor. Os avanços no conhecimento e diagnóstico da FC oferecem esperança ao paciente, mas são necessárias abordagens integradas e suporte multidisciplinar para otimizar os resultados clínicos e a qualidade de vida. OLIVEIRA RUIZ, CALSAVERINI LEAL, 2023 Desafios e Avanços na Fibrose Cística – Uma Revisão Bibliográfica Moduladores de CFTR, percussão, drenagem postural. Redução da adesão ao tratamento devido complexidade e acesso limitado a serviços. MALAQUIAS, CARDOSO, 2023 Desafios no manejo da Fibrose Cística Fisioterapia respiratória (Drenagem postural, vibração pulmonar manual, técnica de expiração forçada, drenagem autógena). Antibióticos e Psicoterapia. A fisioterapia respiratória e o suporte psicológico são essenciais para o tratamento. NIEHAMMER et al., 2022 Cough suppression and HRQoL in adult people with cystic fbrosis: an unexplored correlation Questionário “Cystic Fibrosis Questionnaire-Revised (CFQ-R+14) e questionário sobre supressão da tosse desenvolvido pela equipe de pesquisa. A supressão da tosse, impacta negativamente a qualidade de vida. MENEZES et al., 2022 Atuais técnicas de intervenções fisioterapêuticas para remoção de secreção em crianças diagnosticadas com Fibrose Cística: uma revisão integrativa de literatura Expiração lenta e prolongada (ELPr), aumento do fluxo expiratório (AFE), hiperinsuflação manual, drenagem autógena, aspiração traqueal, expiração lenta total com a glote aberta em decúbito lateral (ELTGOL), vibrocompressão. É indispensável a assistência do fisioterapeuta para que as técnicas de remoção de secreção sejam devidamente aplicadas, visando a melhora da oxigenação e da qualidade de vida. GURSES et al., 2022 Does the effect of comprehensive respiratory physiotherapy home‐program differ in children with cystic fibrosis and non‐cystic fibrosis bronchiectasis? Respiração diafragmática, expansão torácica, flutter ou shaker, drenagem postural com percussões, técnicas de tosse e aconselhamento sobre atividade física. Fisioterapia respiratória é benéfica para função pulmonar e força muscular. MACIEL et al., 2021 3 evidências da fisioterapia respiratória no tratamento da Fibrose Cística Drenagem postural, pressão expiratória positiva (PEP), compressão torácica de alta frequência. A fisioterapia respiratória mostrou-se benéfica e de grande influencia sobre a sintomatologia e qualidade de vida dos portadores de FC. OLIVEIRA et al., 2021 Recursos fisioterapêuticos e aspectos clínicos em pacientes com Fibrose Cística. Tapotagem, vibração, drenagem postural, técnica de expiração forçada (TEF), huffing, oscilação oral de alta frequência, drenagem autógena, ciclo ativo de respiração. Fisioterapia respiratória melhora a qualidade de vida e reduz riscos de infecções pulmonares. DUTRA et al., 2020 Revisão sistemática da literatura sobre a fibrose cística e dados sobre a doença no Brasil. Abordagens sobre genética, fisiopatogenia, microbiologia das infecções pulmonares, manifestações clínicas, critérios clínicos e laboratoriais dos diagnósticos, tratamento e prognóstico. Mudanças na atuação dos profissionais e incorporação de novos recursos tem aumentado a expectativa de vida. MUCHA et al., 2020 Força muscular respiratória e qualidade de vida em crianças e adolescentes com fibrose cística. Avaliação antropométrica, forca muscular respiratória (PImax/PEmax), questinário Cystic Fibrosis Questionnaire, questionário para a qualidade de vida para crianças e pais ou responsáveis. A FC impacta a qualidade de vida, afetando a força muscular respiratória. MELLO et al., 2020 Perfil epidemiológico e social de crianças e adolescentes com fibrose cística Perfil sociodemográfico e adesão ao tratamento fisioterapêutico, nebulização e prática de atividade física. A falta de condições econômicas impacta a vigilância necessária. SÁNCHEZ-AZOFRA et al., 2020 Cystic fibrosis outpatient treatment and medical costs: a retrospective analysis Avaliação do custo farmacêutico e médico. A FC é uma condição dispendiosa para o sistema de saúde. STEFFEN et al., 2019 Upper Airway Findings and Markers of Lung Disease Progression in Patients with Cystic Fibrosis Descrever os achados nasais e patógenos mais comuns na FC e investigar associação entre os achados e a progressão da doença. O swab nasal foi positivo para Staphylococcus aureus, Pseudomonas aeruginosa, Pseudomonas cepacea, Stenotrophomonas maltophila. CHENEY et al., 2020 Health-related quality-of-life in children with cystic fibrosis aged 5-years and associations with health outcomes Avalição da QV utilizando os questionários Pediatric Quality of Life Inventory (PedsQLTM) e Cystic Fibrosis Questionnaire-Revised (CFQ-R). Comparação da terapia por BAL versus terapia padrão. Não houve diferenças nas terapias, e crianças com fibrose cística relataram pior qualidade de vida relacionada a saúde.
Fonte: Autoria própria, 2024.
4 DISCUSSÃO
A FC é uma doença grave e potencialmente letal, as infecções pulmonares especialmente causadas por Staphylococcus aureus podem levar à morte prematura por isso é essencial o conhecimento do perfil microbiológico dos pacientes (BARBOSA, et al. 2024). Steffen, et al. (2019) descreveram os achados e patógenos encontrados nas vias áreas superiores sendo eles Staphylococcus aureus, Pseudomonas aeruginosa, Pseudomonas cepacea, Stenotrophomonas maltophila descritos como causadores de infecções pulmonares crônicas.
Por afetar diretamente o contexto biopsicossocial dos pacientes, a FC exige um cuidado contínuo e multidisciplinar, que vise não apenas a estabilização clínica, mas também o fortalecimento do bem-estar psicológico, emocional e social dos pacientes. A interação entre a equipe de saúde e os familiares desempenha um papel crucial na adesão ao tratamento (DUARTE et al. 2022).
Conforme relatado no estudo de Silva; Souto (2024) o teste do suor é uma ferramenta fundamental no diagnóstico precoce e no monitoramento da progressão da FC. Além do teste do suor como padrão ouro Ribeiro et al. (2021) relataram a importância do teste do pezinho, da anamnese e do sequenciamento genético como ferramentas diagnósticas e norteadoras para as decisões terapêuticas que serão realizadas durante o tratamento.
Após utilizar o questionário Cystic Fibrosis Questionnaire-Revised (CFQ-R) para avaliar a qualidade de vida relacionada à saúde (QVRS), Grysztar et al. (2024), verificaram que as exacerbações respiratórias têm um impacto significativo na mesma, reforçando que as exacerbações frequentes podem levar a uma deterioração na saúde física e mental dos pacientes, mesmo quando tratados com terapias modernas.
A introdução das terapias moduladoras do CFTR, conforme abordado em Santos et al. (2024), representou um marco no tratamento da fibrose cística. Contudo, persistem desafios, como a variabilidade nas respostas aos moduladores e questões relacionadas aos custos e à acessibilidade, que podem influenciar os resultados terapêuticos. O estudo de Lopes (2024), demonstra que o avanços das terapias moduladoras de CFTR tem revolucionado o tratamento da FC, mas o impacto pleno dessas terapias ainda depende de sua acessibilidade global e da adaptação dos tratamentos às diversas mutações genéticas que causam a doença, tornando seu manejo complexo. Segundo Upadhyay, et al. (2024) entender as causas celulares e moleculares da doença facilitam a criação de tratamentos que visam as disfunções subjacentes causadas pelas mutações do CFTR, colaborando para melhorar a qualidade de vida.
A análise de Lanfranchi et al. (2024) sobre as dificuldades em compreender as exacerbações respiratórias em pacientes tratados com moduladores do CFTR destaca uma questão relevante dessa abordagem terapêutica, a complexidade em monitorar e identificar exacerbações respiratórias em pacientes que estão mais estáveis devido ao tratamento. Essas podem não ser facilmente perceptíveis ou se apresentar de forma atípica, o que torna o acompanhamento mais desafiador para os profissionais de saúde, podendo ocasionar agravos na progressão da doença.
As terapias respiratórias têm mostrado efeitos positivos na qualidade de vida dos pacientes com FC. De acordo com Gurses, et al. (2022); Oliveira et al. (2021) e Maciel et al. (2021), a prática contínua de fisioterapia respiratória alivia os sintomas da doença, reduz queixas e proporciona bem-estar geral dos pacientes. Segundo Mucha et al. (2020) a FC pode ocasionar a redução da força respiratória, o fortalecimento da musculatura respiratória é essencial pois está ligado a uma maior capacidade funcional, tolerância ao exercício físico e preservação da qualidade de vida.
Conforme descrito por Malaquias; Cardoso (2023) e Niehammer, et al. (2022), técnicas como drenagem postural, vibração pulmonar manual e expiração forçada são fundamentais para a remoção de secreções pulmonares, prevenindo e controlando exacerbações da doença. Menezes, et al. (2022) ressaltam a importância da aplicação adequada das técnicas realizadas por profissionais capacitados para a remoção de secreções, melhorando significativamente a função pulmonar e a qualidade de vida dos pacientes.
Segundo Reis; Neves (2024) as terapias respiratórias são eficazes a curto prazo para melhorar a função pulmonar e reduzir exacerbações durante episódios de infecção. Cheney et al. (2020) destacam que, apesar da aplicação das terapias respiratórias, crianças com FC continuam a relatar uma qualidade de vida relacionada à saúde significativamente mais baixa.
Alguns fatores são condicionantes, e conforme mencionado por Mello, et al. (2020), a falta de condições econômicas pode levar a uma vigilância inadequada e ao comprometimento da adesão ao tratamento, impactando negativamente a evolução da doença e a qualidade de vida dos pacientes. Sánchez-Azofra, et al. (2020) complementam esse ponto ao destacarem os elevados custos do tratamento da FC, que impõem um fardo adicional sobre as famílias e os sistemas de saúde.
A adesão ao tratamento continua sendo um desafio conforme apontado por Oliveira; Ruiz; Calsaverini Leal (2023), a complexidade dos regimes terapêuticos, combinada com a dificuldade de acesso a serviços especializados, pode reduzir a adesão dos pacientes ao tratamento, especialmente em áreas com recursos limitados. Dainavas, et al. (2024) confirmam essa relação ao correlacionar a maior renda familiar com melhores resultados na qualidade de vida, destacando como fatores socioeconômicos podem influenciar diretamente a gestão da doença e a percepção de bem-estar.
O sucesso do tratamento está diretamente relacionado à habilidade do profissional em reconhecer e ajustar abordagens que favoreçam tanto o bem-estar físico quanto emocional do paciente e de seus familiares (PARAZZI, 2019). Dutra et al. (2020) ressaltam que a expectativa de vida dos pacientes vem aumentando significativamente devido a incorporação de novas tecnologias para diagnóstico e tratamento, exigindo mudanças na atuação dos profissionais e incorporação de novos recursos terapêuticos para o manejo da FC.
5 CONCLUSÃO
As terapias respiratórias, quando aplicadas por profissionais capacitados, são eficazes para a melhora da função pulmonar e redução de exacerbações durante episódios de infecções pulmonares e melhora da qualidade de vida. Dentre as terapias destacam-se a drenagem postural, pressão expiratória positiva, compressão torácica de alta frequência, tapotagem, vibração, vibrocompressão, técnica de expiração forçada, huffing, oscilação oral de alta frequência, drenagem autógena, ciclo ativo de respiração, expiração lenta e prolongada, aumento do fluxo expiratório, hiperinsuflação manual, aspiração traqueal, expiração lenta total com a glote aberta em decúbito lateral.
A FC é uma doença complexa que exige cuidados multidisciplinares. Pesquisas futuras devem explorar estratégias para otimizar a eficácia dessas terapias, garantindo acessibilidade e melhora da qualidade de vida para todos os pacientes.
REFERÊNCIAS
BASILE, Melissa et al. The impact of elexacaftor/tezacaftor/ivacaftor on cystic fibrosis health-related quality of life and decision-making about daily treatment regimens: a mixed methods exploratory study. Therapeutic Advances in Chronic Disease, v. 15, p. 20406223241264477, 2024.
CHENEY, Joyce et al. Health-related quality-of-life in children with cystic fibrosis aged 5-years and associations with health outcomes. Journal of Cystic Fibrosis, v. 19, n. 3, p. 483-491, 2020.
DAINAVAS, Dimitris et al. The impact of cost of living on the quality of life of cystic fibrosis patients: a study in Greece. Cureus, v. 16, n. 6, 2024.
DICKINSON, K. M.; COLLACO, J. M. Cystic fibrosis. Pediatrics in Review, v. 42, n. 2, p. 55-67, fev. 2021. DOI: 10.1542/pir.2019-0212. PMCID: PMC8972143.
DUTRA, Natália Alves et al. Revisão sistemática da literatura sobre a fibrose cística e dados sobre a doença no Brasil. Anais do Seminário Científico do UNIFACIG, n. 6, 2020.
HUMAJ-GRYSZTAR, Magdalena; RACHEL, Marta; BONIOR, Joanna. Polish cystic fibrosis patients’ health-related quality of life and its influencing factors: a cross-sectional, single-centre study. In: Healthcare. MDPI, 2024. p. 1183.
LANFRANCHI, Chiara et al. A case of severe pulmonary exacerbation in a young patient with cystic fibrosis in the era of CFTR modulators. International Journal of Infectious Diseases, v. 147, p. 107190, 2024.
LOPES, Lorenzo de Barros et al. FIBROSE CÍSTICA: PATOGÊNESE, DIAGNÓSTICO E INOVAÇÕES TERAPÊUTICAS. Revista Ibero-Americana de Humanidades, Ciências e Educação, v. 10, n. 8, p. 2403-2409, 2024.
MACIEL, Júlia Maria de Sousa; SALES, Weslley; BARBOSA, Renata Ramos Tomaz. Evidências da fisioterapia respiratória no tratamento da fibrose cística. Scientia: Revista Científica Multidisciplinar, v. 6, n. 2, p. 41-60, 2021.
MALAQUIAS, Soraya Beatriz Paula; CARDOSO, Alessandra Marques. Desafios no manejo da fibrose cística. Revista Brasileira Militar de Ciências, v. 9, n. 23, 2023.
MALL, M. A. et al. Cystic fibrosis. Nature Reviews Disease Primers, v. 10, n. 1, p. 53, 8 ago. 2024. DOI: 10.1038/s41572-024-00538-6.
Manual MSD. CYSTIC FIBROSIS FOUNDATION. Bethesda, Maryland: Cystic Fibrosis Foundation, 2022. Disponível em: https://www.msdmanuals.com/pt/profissional/pediatria/fibrose-c%C3%ADstica/fibrose-c%C3%ADstica. Acesso em: 16/11/2024.
MENEZES, Thainá Barros de et al. Atuais técnicas de intervenções fisioterapêuticas para remoção de secreção em crianças diagnosticadas com fibrose cística: uma revisão integrativa de literatura. Brazilian Journal of Health Review, v. 5, n. 6, p. 22075-22087, 2022.
MELLO, Marcos Filipe da Silva et al. Perfil epidemiológico e social de crianças e adolescentes com fibrose cística. Revista Educação em Saúde, v. 8, n. 1, p. 151-160, 2020.
MINISTÉRIO DA SAÚDE. Fibrose cística atinge 1 a cada 10 mil nascidos vivos no Brasil. 2022. Disponível em: https://www.gov.br/ebserh/pt-br/comunicacao/noticias/fibrose-cistica-atinge-1-a-cada-10-mil-nascidos-vivos-no-brasil. Acesso em: 18 nov. 2024.
PARAZZI, Paloma. Monitorização da adesão e ajustes terapêuticos. ASSOBRAFIR Ciência. Recomendação Brasileira de Fisioterapia na Fibrose Cística: Um guia das boas práticas clínicas. Volume 10. Suplemento 1. 2019.
PEREIRA, Táylla Fernanda dos Santos. Fisioterapia respiratória na fibrose cística: técnicas e recursos utilizados em um serviço de referência. 2023.
REIS, João Victor Lopes; NEVES, Camila Danielle Cunha. Terapias de higiene brônquica durante exacerbações de pacientes com fibrose cística: uma revisão de literatura. 2024.
RIBEIRO, Maria Natália Alves et al. Fibrose cística: histórico e principais meios para diagnóstico. Research, Society and Development, v. 10, n. 3, p. e11710313075-e11710313075, 2021.
RUIZ, Talicya Renata Oliveira; LEAL, Renata Calsaverini. Desafios e avanços na fibrose cística: uma revisão bibliográfica. Revista Corpus Hippocraticum, v. 2, n. 1, 2023.
SANTOS, Luiz Henrique Cunha dos et al. Perspectivas futuras em terapias moduladoras para fibrose cística: avanços e desafios. Research, Society and Development, v. 13, n. 5, p. e0613545653-e0613545653, 2024.
SHADI, Danial et al. Protocol: development and validation of a supportive programme for family caregivers of children suffering from cystic fibrosis: protocol for a sequential exploratory mixed-methods study. BMJ Open, v. 14, n. 6, 2024.
SILVA, Ana Beatriz Gonçalves da. Fibrose cística: principais características clínicas e métodos de diagnóstico. 2024.
UPADHYAY, Kirti et al. Current and future therapeutic approaches of CFTR and airway dysbiosis in an era of personalized medicine. Journal of Family Medicine and Primary Care, v. 13, n. 6, p. 2200-2208, 2024.
VENEZIANO, Leonardo Squinello Nogueira et al. Recursos fisioterapêuticos e aspectos clínicos em pacientes com fibrose cística. Revista Científica da Faculdade Quirinópolis, v. 1, n. 11, p. 17-33, 2021.
ZAMBERLAN, Samantha. Avaliação da adesão ao tratamento medicamentoso em crianças e adolescentes com fibrose cística e fatores clínicos e nutricionais associados. 2023.
1 Discente do curso de Medicina do Centro Universitário de Patos de Minas -UNIPAM.
2 Docente do curso de Medicina do Centro Universitário de Patos de Minas -UNIPAM.