REGISTRO DOI: 10.69849/revistaft/ma10202411191426
Priscila Aquino Pinheiro
Larissa Aquino Pinheiro
Giovanna Aquino Pinheiro
Júlia Aquino Pinheiro
RESUMO
A Síndrome de Yamaguchi é um subtipo de cardiomiopatia hipertrófica caracterizada pela hipertrofia no ápice ventricular esquerdo. É uma síndrome rara no Brasil, sendo comumente encontrada no Japão. Possui um grande espectro clínico, apresentando-se desde paciente assintomáticos a morte súbita. A elucidação diagnóstica se dá por meio de alterações no eletrocardiograma e no ecocardiograma, sendo a ressonância magnética cardíaca o exame mais específico. O tratamento inicial é clínico, podendo estender-se a procedimentos cirúrgicos se refratário ao manejo medicamentoso. O presente estudo tem como objetivo apresentar um caso clínico de Síndrome de Yamaguchi em um paciente do estado do Ceará, enfatizando a importância de incluir a síndrome no diagnóstico diferencial da sintomatologia com dor torácica, dispneia e palpitações.
Palavras-Chave: Miocardiopatia Hipertrófica Apical; Dor torácica; Síncope; Coração.
ABSTRACT
Yamaguchi Syndrome is a subtype of hypertrophic cardiomyopathy characterized by hypertrophy in the left ventricular apex. It is a rare syndrome in Brazil, being commonly found in Japan. It has a wide clinical spectrum, ranging from asymptomatic patients to sudden death. Diagnosis is elucidated through changes in the electrocardiogram and echocardiogram, with cardiac magnetic resonance imaging being the most specific exam. The initial treatment is clinical, and may be extended to surgical procedures if refractory to drug management. This study aims to present a clinical case of Yamaguchi Syndrome in a patient in the state of Ceará, emphasizing the importance of including the syndrome in the differential diagnosis of symptoms with chest pain, dyspnea and palpitations.
Keywords: Apical Hypertrophic Cardiomyopathy; Chest Pain; Syncope; Heart.
1. INTRODUÇÃO
A síndrome de Yamaguchi, também conhecida por cardiomiopatia hipertrófica apical (CMHA) é um subtipo raro e de caráter autossômico dominante da cardiomiopatia hipertrófica (CMH). (8) Inicialmente, pela sua descoberta em 1979 no Japão, presumia-se que a patologia estava limitada apenas à população asiática, cuja prevalência atual é de 13-14%. Entretanto, estudos recentes já evidenciam casos em outras nações, apresentando prevalência de 3% nos Estados Unidos. (9)
Comumente, o tipo apical da CMH envolve apenas o ventrículo esquerdo (VE), mais raramente, compromete o ventrículo direito ou ambos. Apresenta principalmente hipertrofia localizada em ápice do VE, diferente dos demais subtipos, que acometem sobretudo o septo ventricular. (9)
A CMHA incide principalmente no sexo masculino, na faixa etária de 40 a 50 anos (2, 4). Apresenta amplo espectro clínico, variando desde quadros assintomáticos à presença de dispneia, dor torácica, palpitações, e até mesmo a complicações como arritmias malignas, infarto e morte súbita. (4, 7).
Frequentemente é diagnosticada incidentalmente por meio de exames como o eletrocardiograma (ECG) e o ecocardiograma transtorácico (ECOTT), evidenciando, sobretudo, a inversão de onda T gigante e sinais de hipertrofia do ventrículo esquerdo respectivamente. (8) Entretanto, diversas vezes, mais exames são necessários ao diagnóstico, como a ressonância magnética e a ventriculografia, porém este último tem limitação por seu caráter invasivo. (2)
Diante da apresentação clínica heterogênea do paciente e por, muitas vezes, o quadro simular uma síndrome coronariana aguda (SCA), a CMHA frequentemente é subdiagnosticada ou apresenta diagnóstico tardio, corroborando a um maior risco para desenvolver complicações graves.
O tratamento do subtipo apical consiste em controle sintomático a depender da clínica do doente, podendo em casos refratários ser necessário procedimento cirúrgico. Seu prognóstico é relativamente benigno, com menor mortalidade em relação aos demais subtipos, entretanto, não exclui a necessidade de acompanhamento rigoroso a fim de prevenir complicações com risco à vida. (9)
Diante da variabilidade clínica que o quadro pode apresentar e da morbimortalidade cardiovascular atribuída às complicações dessa patologia, o presente estudo tem como objetivo enriquecer o conhecimento médico acerca dessa síndrome.
2. OBJETIVOS
2.1. Objetivo Geral
Relatar o caso de um paciente do sexo masculino, 70 anos, com o diagnóstico de Síndrome de Yamaguchi, sendo realizado seguimento com terapêutica medicamentosa.
2.2. Objetivos Específicos
- Relatar o caso clínico, métodos diagnósticos e tratamento realizado.
- Definir e descrever o quadro clínico da Síndrome de Yamaguchi.
- Relatar os métodos diagnósticos e os princípios terapêuticos da Síndrome de Yamaguchi.
3. MÉTODOS
3.1. Tipo do estudo
O presente estudo trata-se de um Relato de Caso Clínico acompanhado de revisão da literatura sobre a Síndrome de Yamaguchi, sua apresentação clínica, abordagem diagnóstica e proposta terapêutica.
3.2. Local do estudo
O estudo foi realizado com um paciente que esteve internado em dezembro de 2020 e em junho de 2021 no Hospital de Messejana Dr. Carlos Alberto Studart Gomes (HM). O serviço caracteriza-se por ser um hospital de atenção terciária, o qual promove a assistência à população do Ceará através do Sistema Único de Saúde (SUS), nas áreas cardiovascular, torácica e pulmonar, além de atuar como centro de ensino e pesquisa.
A unidade de estudo deu-se na enfermaria da Unidade B, local onde o paciente esteve internado e onde foram coletados os dados durante a revisão do prontuário.
3.3 População e amostra
Paciente do sexo masculino, 70 anos, com quadro clínico de síncope associada a palpitações, dispneia e dor torácica, sendo, posteriormente, diagnosticado com Síndrome de Yamaguchi.
3.4. Riscos e Benefícios
Os riscos que podemos citar neste estudo são a exposição do coletor dos dados e risco de identificação do paciente. Nenhum dado que possa identificar o paciente foi ou será divulgado.
Os benefícios ao relatar este caso são discutir sobre a abordagem de uma condição pouco diagnosticada, alertando para a importância de gerar suspeita desta síndrome em casos de síncope, e compartilhar os resultados após a instituição do tratamento adequado.
3.5. Coleta de dados
Os dados foram obtidos pela coleta da história clínica e da revisão do prontuário médico de um paciente diagnosticado com Síndrome de Yamaguchi, que esteve internado no Hospital de Messejana Dr. Carlos Alberto Studart Gomes em dezembro de 2020 e em junho de 2021. A revisão de literatura foi realizada utilizando-se das plataformas científicas PubMed e ScienseDirect. O escopo do estudo foi realizado usando a ferramenta do Microsoft Word.
4. RELATO DO CASO CLÍNICO
Paciente do sexo masculino, 70 anos, tabagista (100 maços x ano), portador de Hipertensão Arterial Sistêmica (HAS), dislipidemia e hipotireoidismo acompanhado em ambulatório de cardiologia. Relata que, em 2016, apresentou episódio de síncope associado a palpitação e sudorese. Procurou atendimento médico, onde foi realizado cateterismo cardíaco com angioplastia e colocação de stent.
Desde então, refere quadros semelhantes esporadicamente (um episódio ao ano), com resolução espontânea. Em dezembro de 2020, apresentou novo quadro de síncope precedida por palpitações, dispneia e sudorese profusa, sintomas que duraram alguns minutos e melhoraram espontaneamente sem intervenção terapêutica. Em novembro de 2020, apresentou novo episódio semelhante com perda da consciência. Devido a piora progressiva do quadro clínico, procurou atendimento médico ambulatorial, onde foi encaminhado à emergência do Hospital de Messejana. Admitido na emergência estável clinicamente, eupneico em ar ambiente, sem queixas clínicas, exames laboratoriais todos dentro da normalidade, exceto por alteração em função tireoidiana. Optado pelo internamento hospitalar para investigação do caso.
Realizou Eletrocardiograma (ECG) (imagem 1) que mostrou sinais de sobrecarga do ventrículo esquerdo, com alterações de repolarização. Após, realizou Ecocardiograma Transtorácico (ECOTT) que evidenciou aumento moderado do átrio esquerdo (ViAE = 45 ml/m²). Presença de hipertrofia do ventrículo esquerdo, predominantemente em região apical, sem gradiente médio-apical ou em via de saída do VE. Função sistólica do ventrículo esquerdo preservada. FEVE = 65% (Teicholz). Valva aórtica trivalvular espessada, com pontos de calcificação, abertura e mobilidade preservadas, sem gradiente sistólico significativo ou refluxo. Valva mitral espessada, com ponto de calcificação no anel mitral, com abertura e mobilidade preservadas, apresentando refluxo discreto. Valva tricúspide com boa abertura e mobilidade, apresentando refluxo leve. Pressão de saída da Artéria Pulmonar (PSAP) estimada em 26 mmHg. Valva pulmonar com boa abertura e mobilidade, sem gradiente sistólico significativo e com refluxo discreto. Exame compatível com miocardiopatia hipertrófica (Yamaguchi). Realizou Holter que evidenciou ritmo de base sinusal, extrassístoles ventriculares raras, com um episódio de Taquicardia Ventricular não sustentada (TVNS) de 6 batimentos, extrassístoles atriais raras, intervalo PR em torno de 0,13 segundo.
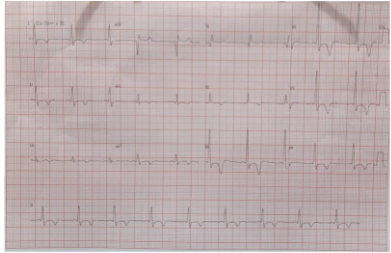
Imagem 1: ECG evidenciando ritmo sinusal. Sobrecarga do ventrículo esquerdo. Infradesnivelamento do Segmento ST em parede lateral anterolateral. Inversão de onda T em parede anterolateral.
Realizado cateterismo cardíaco que mostrou Tronco da Coronária Esquerda (TCE) e Descendente Anterior (DA) sem lesões; Diagonal de moderada importância com lesão 50-60% no óstio e terço proximal e lesão suboclusiva (90%) no terço médio; Circunflexa sem lesão; 1° Marginal de grande importância e com lesão 40% no terço distal; 2° marginal sem lesão; Coronária Direita dominante com stent pérvio, sem lesão; Diagonal Posterior e Ventricular Posterior sem lesão. Realizado angioplastia da diagonal com sucesso.
Iniciado tratamento medicamentoso com bisoprolol (5mg/dia), losartana (50mg/dia), atorvastatina (40mg/dia), AAS (100mg/dia), clopidogrel (75mg/dia) e levotiroxina (12,5mcg/dia). Evoluiu ao longo do internamento com melhora clínica e assintomática, optou-se por alta hospitalar com solicitação de Ressonância Magnética cardíaca ambulatorial, orientação para uso das medicações descritas e retorno ao ambulatório com resultado de exame.
Em junho de 2021, apresentou novo episódio de síncope com perda da consciência durante a madrugada, associado à sudorese e à dor torácica em aperto, com irradiação para mandíbula. Procurou atendimento médico no Hospital de Messejana, portando resultado de RNM cardíaca (imagem 2), constando câmaras cardíacas de dimensões preservadas. Função sistólica biventricular normal, FEVE 77%. Massa VE 250g ou 147g/m2. Hipertrofia miocárdica de regiões medianas e apicais do VE, com predomínio apical e espessura máxima de 24mm. Ausência de discinesia ou dissincronia do VD. Presença de fibrose miocárdica nos segmentos mediais e apicais do VE (massa de fibrose de 15g ou 6% da massa ventricular). Optado por novo internamento hospitalar com manutenção das medicações em uso.
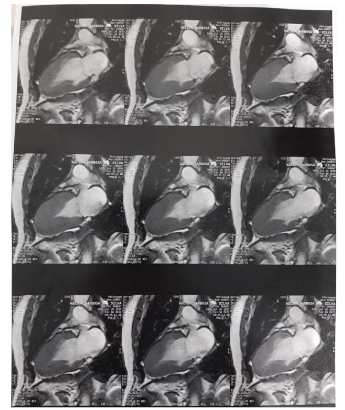
Imagem 2: RNM do coração, realizada em fevereiro de 2021.
Realizado Holter que evidenciou frequência cardíaca (FC) mínima 47 bpm; Extrassístoles ventriculares (ESV) raras, Extrassístoles atriais (ESA) raras, com epiśodicas taquicardias atriais não sustentadas. Intervalo PR 0,13 segundo e Complexo QRS normal. Realizou ECOTT que mostrou aumento do átrio esquerdo, hipertrofia do VE, hipocinesia difusa do VE e disfunção sistólica leve do VE. FEVE 46%. Disfunção diastólica do VE grau 1, PSAP 40 mmHg; valva mitral refluxo leve; valva tricúspide refluxo leve. Optado por alta hospitalar com acompanhamento ambulatorial e uso das medicações prévias.
Paciente evolui com controle importante dos sintomas com adesão terapêutica, sem novos episódios de internamento hospitalar, e mantendo acompanhamento ambulatorial de cardiologia no Hospital de Messejana.
5. DISCUSSÃO
A Síndrome de Yamaguchi, também conhecida como cardiomiopatia hipertrófica apical, é uma forma rara de cardiomiopatia hipertrófica não obstrutiva localizada no ventrículo esquerdo. (7) Foi descrita pela primeira vez em 1979 por Yamaguchi no Japão, local onde a doença tem sua maior prevalência (15%). No entanto, nos últimos anos, vem sendo relatados casos em diversos países, sendo o principal nos Estados Unidos, com uma prevalência de 3%. No Brasil foram encontrados alguns relatos de casos, mas sem estudos fidedignos com cálculo da prevalência. (8)
A síndrome acomete principalmente pacientes do sexo masculino na quarta década de vida. O paciente relatado aqui é do sexo masculino e foi diagnosticado com 70 anos, o que confere um retardo no diagnóstico clínico mesmo com sintomas clínicos desde os 65 anos. Apesar de seu caráter genético autossômico dominante, observa-se menos pacientes com história familiar positiva, sugerindo que fatores ambientais também desempenham papel na doença. (4, 8)
No geral, mais de 50% dos pacientes não apresentam sintomas, sendo o diagnóstico muitas vezes feito de forma incidental. Os pacientes sintomáticos apresentam sintomas leves a graves, como dor torácica, dispneia, palpitações, fadiga e síncope. Alguns casos podem apresentar morte súbita. (7, 8)
Na avaliação clínica, pode ser evidenciada quarta bulha audível e palpável, corroborando com comprometimento do VE. Além disso, cerca de 40% dos pacientes com CMHA apresentam alterações na valva mitral, podendo haver sinais de regurgitação mitral também ao exame. (2, 4)
Diante da variabilidade clínica da doença, deve-se atentar aos diagnósticos diferenciais, principalmente causas secundárias de hipertrofia ventricular esquerda, como estenose aórtica valvar ou subvalvar, hipertensão arterial e cardiomiopatia infiltrativa. (2, 10) Outras patologias inseridas no espectro diferencial da CHMA incluem trombo apical do ventrículo esquerdo, tumores cardíacos apicais, endomiocardiofibrose, compactação ventricular isolada e, principalmente, doença arterial coronariana. (9)
Frequentemente pacientes com subtipo apical podem apresentar quadro clínico que mimetiza uma síndrome coronariana aguda, levando usualmente a uma conduta médica invasiva precoce. (7)
Habitualmente a Síndrome de Yamaguchi é identificada acidentalmente. A suspeita clínica inicia-se por meio do ECG evidenciando sinais típicos do quadro, como ondas T inversas gigantes (com amplitude ≥10 mm) nas precordiais e alta voltagem do complexo QRS na ausência de doença coronariana. Tais achados podem ser encontrados em 11 a 47% dos casos. Estudos recentes relatam que a ausência de ondas T invertidas na síndrome estavam mais associadas à hipertrofia apical localizada em áreas distais, enquanto a presença de tal achado associa-se mais frequentemente a hipertrofia difusa do ápice. (3, 9)
A elucidação diagnóstica é feita por meio de exames de imagem com a evidência de hipertrofia apical, sendo o ecocardiograma o exame de escolha. A visualização de uma espessura assimétrica da parede do VE superior a 15 mm durante a diástole ou valor inferior a 13 mm, mas com clínica, imagem e história familiar positivas, determina o diagnóstico. Outros achados possíveis ao exame são a obstrução do ventrículo médio e a obliteração da cavidade durante a sístole. A alteração persistente também durante a diástole é indicativa de pior prognóstico, com maior risco para embolia sistêmica e arritmias. (3, 9)
Contudo, tais achados podem não ser visualizados de forma fidedigna. Nesse contexto, a ressonância magnética representa uma ferramenta diagnóstica mais confiável, podendo inclusive ser usada para estratificação de risco para morte súbita. Os critérios diagnósticos na RM são espessura da parede apical maior que 15 mm e/ou relação espessura apical do ventrículo esquerdo e da parede basal ≥ 1,3-1,5. (7) Estudos mostram que uma espessura de parede maior ou igual a 30 mm está associada a um risco grave para morte súbita. Ademais, ao exame também pode ser visto um realce tardio pelo gadolínio, compatível com deposição de matriz extracelular e fibrose, resultado de múltiplos episódios de isquemia em microvasculatura. (9)
Outro exame citado no contexto de CMHA é o ventriculograma, porém diante do seu caráter invasivo e pouco acessível está em desuso. Nesse exame é possível visualizar hipertrofia assimétrica no ápice do ventrículo esquerdo, causando uma configuração em “ás de espadas” ao final da diástole. (3, 6, 7)
Confirmado o diagnóstico, a CMHA é tratada clinicamente na maioria dos casos. Mudança no estilo de vida é o primeiro pilar do tratamento, devendo-se priorizar atividade aeróbica de baixa intensidade e evitar algumas exposições, como atividade física extenuante, consumo de álcool e de substâncias psicoativas, temperaturas extremas e desidratação, pois são considerados fatores precipitantes de síncopes ou arritmias (2, 5).
O tratamento medicamentoso inicial deve ser feito com uso de Betabloqueadores, os quais atuam diminuindo a resposta cronotrópica do coração, reduzindo a demanda de oxigênio e maximizando o enchimento diastólico. Não foram encontrados estudos comparando os diferentes tipos de betabloqueadores, mas a subclasse beta-1 seletiva deve ser priorizada para evitar a piora do gradiente médio ventricular (5, 9). O uso de Inibidores da enzima conversora de angiotensina (IECA) e de Bloqueadores dos Canais de Cálcio (BCC) também é recomendado visando controle da frequência cardíaca e redução da pós carga ventricular esquerda, reduzindo a remodelação cardíaca e a fibrose observadas na CMHA. (1, 6)
Pacientes sem melhora clínica, com persistência dos sintomas e permanecendo na classificação 3 ou 4 da NYHA, devem ser avaliados para intervenção cirúrgica. As opções terapêuticas são miectomia estendida com reconstrução ventricular ou miectomia apical. Ao final do procedimento cirúrgico, a espessura ideal da parede é de 10 a 12 mm. Em último caso, está indicado transplante cardíaco sem sintomas refratários às medidas anteriormente explicitadas. (5, 9)
A CMHA apresenta um prognóstico benigno a longo prazo quando comparada com outras formas de cardiomiopatia hipertrófica (6, 8). No entanto, 25% dos pacientes podem evoluir com morbidades cardiológicas, como insuficiência cardíaca, dor torácica, fibrose apical, aneurismas apicais, fibrilação atrial e taquicardia ventricular (2, 3). As características associadas a um mau prognóstico incluem: idade jovem, história familiar de morte súbita e classe NYHA 2, 3 ou 4.
Devido ao risco de morte súbita, é recomendado o uso de cardiodesfibriladores implantáveis (CDIs) como prevenção primária de morte súbita nos seguintes casos: 1) história familiar de morte súbita cardíaca; 2) síncope sem causa aparente; 3) taquicardia ventricular não sustentada assintomática; 4) resposta anormal da pressão arterial ao exercício; e 5) espessura da parede ventricular esquerda maior que 30 mm. (2)
Os pacientes com diagnóstico CMHA devem ser acompanhados anualmente para avaliação clínica e repetição do ecocardiograma (9). Além disso, o acompanhamento dos familiares de primeiro grau é recomendado, visando a identificação precoce da doença. (8)
6. CONCLUSÃO
O relato de caso deste paciente demonstra a importância na suspeição clínica da Síndrome de Yamaguchi em pacientes que apresentam sintomas como dor torácica, dispneia e palpitações. O diagnóstico em momento oportuno viabiliza o tratamento precoce desta síndrome, possibilitando uma melhor qualidade de vida, evitando a recorrência do quadro e reduzindo o risco de complicações secundárias em curto e longo prazos.
REFERÊNCIAS
- ABUGROUN, Ashraf; et al. Apical Hypertrophic Cardiomyopathy: A Case Report. Cardiol Res. Chicago, USA. Vol 8, n 5, p 265-268, october, 2017
- BUITRAGO-GÓMEZ, Nathalia; et al. Apical hypertrophic cardiomyopathy. Yamaguchi syndrome. Acta Médica Colombiana. Cali-Colômbia, Ed. 46, n 4, october, 2021.
- ERIKSSON, Maria; et al. Resultado de longo prazo em pacientes com cardiomiopatia hipertrófica apical. J Am Coll Cardiol. Toronto-Canadá, v. 39, n. 4, pag. 638-645, february, 2002.
- HUGHES, Rebecca; et al. Cardiomiopatia hipertrófica apical: A variante menos conhecida. J AmHeart Assoc. v. 9, n 5, march, 2020.
- JAN, Maria; et al. Cardiomiopatia hipertrófica apical: estado atual. Int J Cardiol. Milwaukee-Wisconsin, v. 222, pag. 745-759, july, 2016.
- PAYUS, Alvin; et al. Síndrome de Yamaguchi uma síndrome coronária pseudoaguda do jovem: Relato de caso sobre cardiomiopatia hipertrófica apical. J Med Sci. Sabah-Malaysia, v. 39, n. 4, pag. 197-199, august, 2019.
- RUHELA, Manish; et al. Yamaguchi syndrome: A mimicer of acute coronary syndrome. Journal of Family Medicine and Primary Care. Rajastha-India, Vol 11, n 5, p. 2223 – 2225, mayo, 2022.
- SALAMÉ, Cecilio; et al. Cardiomiopatia hipertrófica apical (Síndrome de Yamaguchi) em paciente assintomático,não asiático, diagnosticado por ressonância magnética, um relato de caso. Brazilian Journal of Development. Curitiba- Brasil, Vol. 7, n 9, p. 88339-88345, setembro, 2021.
- SHAH, Farhan; et al. A Novel Case of Yamaguchi Syndrome in a Hispanic Male. Cureus: Journal of Medical Sciense. Vol 13, n 9, january, 2021.
- YUSUF, Sayed; et al. Cardiomiopatia hipertrófica apical. World J Cardiol. Houston-Texas, v. 3, n.9, pag. 256-259, july, 2011.