REGISTRO DOI: 10.5281/zenodo.10582314
Mylena Gaudêncio Bezerra¹, Jeanina Cabral Dionizio¹, Arthur Felipe Barbosa Vasconcelos¹, Paulo Antônio Farias Lucena², Isabella de Araújo Mota²
RESUMO
A doença de Creutzfeldt-Jakob (DCJ) é uma condição neurodegenerativa rara, rapidamente progressiva, não existe uma terapia modificadora da doença e inevitavelmente ocorre um desfecho fatal. É causada pelo acúmulo de proteínas priônicas mal dobradas no tecido cerebral. A apresentação clínica é bastante heterogênea e o tempo médio de sobrevida entre o diagnóstico e o óbito é de alguns meses. Os critérios de diagnóstico clínico utilizam uma combinação dos sintomas neuropsiquiátricos, associados as alterações da ressonância magnética (RM) do crânio e do eletroencefalograma (EEG), assim como a possibilidade de encontrar a proteína 14-3-3 no líquido cefalorraquidiano. O diagnóstico confirmatório é realizado apenas no post mortem através da necropsia. Nos últimos anos surgiu o teste da detecção da amplificação cíclica por enovelamento incorreto das proteínas induzida por tremor (RT-QUIC). Objetiva-se descrever um caso diagnosticado em um hospital terciário da Paraíba, como provável doença de Creutzfeldt-Jakob esporádica.
Palavras-chave: Doença de Creutzfeldt-Jakob, Doença priônica, Encefalopatias
ABSTRACT
Creutzfeldt-Jakob disease (CJD) is a rare neurodegenerative condition that is rapidly progressive, there is no disease-modifying therapy and a fatal outcome inevitably occurs. It is caused by the accumulation of misfolded prion proteins in brain tissue. The clinical presentation is quite heterogeneous and the average survival time between diagnosis and death is a few months. The clinical diagnostic criteria use a combination of neuropsychiatric symptoms associated with alterations in magnetic resonance imaging (MRI) of the skull and electroencephalogram (EEG), as well as the possibility of finding the 14-3-3 protein in cerebrospinal fluid. Confirmatory diagnosis is only made at the post-mortem through necropsy. In recent years, the test for the detection of cyclic amplification by misfolding of proteins induced by tremor (RT-QUIC) has emerged. The aim is to describe a case diagnosed in a tertiary hospital in Paraíba as probable sporadic Creutzfeldt-Jakob disease.
Keywords: Creutzfeldt-Jakob Disease, Prion Disease, Encephalopathies
INTRODUÇÃO
A doença de Creutzfeldt-Jakob (DCJ) é uma doença neurológica rara, dividida em quatro subtipos que incluem DCJ esporádica, genética, iatrogênica e variante. Causada pela partícula chamada de príon, abreviação de “proteinaceous infectious particle”, onde um conjunto da isoforma mal dobrada (PrPSc) da proteína priônica celular codificada pelo hospedeiro (PrPc) está associada a deposição de placas amiloides nas regiões sinápticas e perivasculares, causando lesões espongiformes nas regiões do córtex cerebral, gânglios da base, tálamo e cerebelo resultando em degeneração neuronal.1,2
A DCJ é a encefalopatia espongiforme transmissível mais comum, de rápida progressão, sem proposta terapêutica específica, com um tempo de sobrevida de meses e 100% de mortalidade.3 A incidência mundial descrita é de 1,5 a 2,0 casos novos a cada 1.000.000 de habitantes-ano.4 A apresentação clínica é inespecífica, composta por sintomas de demência rapidamente progressiva, sinais piramidais/extrapiramidais, alterações visuais ou cerebelares e mutismo acinético. Frequentemente associada a sintomas psiquiátricos, insônia e cefaleia. Doença rara com um espectro amplo de sintomas inespecíficos que leva a uma baixa suspeição clínica, possivelmente sendo subdiagnosticada.5 Objetiva-se descrever um caso diagnosticado em um hospital terciário da Paraíba, como provável doença de Creutzfeldt-Jakob esporádica com base nos critérios do CDC de 2018 para DCJ.6
RELATO DO CASO
Mulher, 62 anos, procedente de João Pessoa-PB, três meses antes da internação hospitalar apresentou quadro insidioso de apatia, isolamento social, recusa alimentar e gastrite, diagnosticada com depressão e em acompanhamento psiquiátrico, entretanto, rapidamente evoluiu com perda importante da funcionalidade. Solicitada ressonância magnética (RM) cranioencefálica, obteve-se resultado inespecífico, focos de sinal anômalo esparsos na substância branca dos hemisférios cerebrais, podendo corresponder a microangiopatia degenerativa. Permaneceu com padrão de piora do declínio funcional, totalmente dependente para as atividades básicas diárias, associado ao comprometimento progressivo da marcha e rigidez, sendo transferida para investigação neurológica.
O quadro foi interpretado como demência rapidamente progressiva e a hipótese de DCJ foi aventada. Solicitada nova RM de crânio, ponderada na sequência de difusão (DWI) e T2/FLAIR que evidenciou alteração de sinal acometendo de forma bilateral e simétrica os núcleos da base e de forma assimétrica o córtex dos lobos frontotemporoparietais, mais evidente à esquerda (Figura 1), que são sinais altamente sugestivos da doença. Foi feita coleta de líquido cefalorraquidiano (LCR) para avaliação da presença da proteína 14-3-3, a qual foi confirmada, RT-QUIC detectado, cultura negativa e painel molecular para infecções não identificou nenhum agente infeccioso no LCR. O eletroencefalograma (EEG) registrou acentuada desorganização difusa do ritmo de base, com ondas bifásicas ou trifásicas difusas, intermitentes, com padrão periódico e semiperiódico em uma frequência de 0,5 -1,0 Hz em aproximadamente 50% do traçado registrado, sugestivo da doença. Paroxismos epileptiformes do tipo onda aguda seguida de onda lenta, sobre a região frontotemporal de ambos os hemisférios cerebrais, que ocorreram de forma síncrona e assíncrona, em baixa incidência. A função tireoidiana e a vitamina B12 estavam normais, assim como função hepática, renal, provas reumatológicas e inflamatórias. VDRL, HTLV e HIV foram não reagentes. Tomografias de tórax, abdome e pelve sem alterações. Colonoscopia e endoscopia digestiva sem alterações importantes.
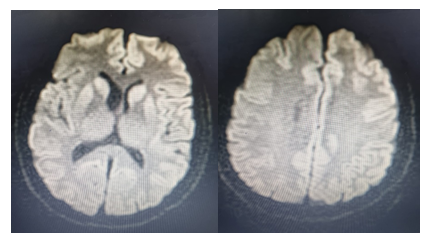
FIGURA 1
RESSONÂNCIA MAGNÉTICA CEREBRAL 3 MESES APÓS O INÍCIO DOS SINTOMAS
Cortes axiais na sequência DWI. Figura da esquerda apresenta hipersinal simétrico nos núcleos da base (núcleos caudados e putâmens). Figura da direita evidência hipersinal cortical dos hemisférios cerebrais nas regiões frontoparietais, mais evidente à esquerda.
A paciente evoluiu com postura distônica de mãos, mioclonias, mutismo acinético, apresentou rigidez generalizada e clonus inesgotável à esquerda, assim como a presença dos reflexos mentoniano, palmo-mentoniano e glabelar inesgotável. Instituído os cuidados paliativos, após 12 dias de internação hospitalar sofreu uma parada cardiorrespiratória e foi à óbito com aproximadamente quatro meses do início dos sintomas. Não houve a confirmação do diagnóstico através dos testes neuropatológicos e/ou imunocitoquímicos, apesar da solicitação ao serviço de verificação de óbito (SVO), foi alegado a impossibilidade da realização da necropsia nesse caso.
DISCUSSÃO
A DCJ familiar foi considerada improvável nesse caso, dada a ausência de história familiar com sintomas semelhantes. A DCJ variante também é improvável, pois apresentou uma duração mais curta da doença e tampouco os achados na RM foram característicos não houve alteração de sinal bilateral nos tálamos. A DCJ iatrogênica foi descartada, pois a paciente não realizou nenhum procedimento neurocirúrgico ou transplante de córnea. Diante das principais manifestações clínicas, o tempo de evolução da doença, os resultados dos exames de RM de encéfalo, EEG, presença no LCR da proteína 14-3-3 e RT QUIC detectado foi firmado o diagnóstico de provável DCJ esporádica.
A solicitação do RT-QUIC pode nortear o diagnóstico da presença de proteína priônica anômala no LCR pre mortem. A Rede Europeia de Vigilância da DCJ realizou um estudo para verificar a concordância entre seus centros na realização do teste do RT-QuIC no LCR, confirmando uma excelente concordância entre os diferentes centros, com um elevado grau de especificidade (99% a 100%) para o diagnóstico de DCJ e recomendou a inclusão do RT- QUIC nos critérios diagnósticos revistos da Rede Europeia de Vigilância da DCJ para DCJ.7
CONCLUSÃO
O relato do caso aponta a necessidade de aumentar a suspeição clínica para DCJ em pacientes com transtornos psiquiátricos associados a uma demência rapidamente progressiva. Também seria importante acrescentar o uso do RT-QuIC na rotina diagnóstica da DCJ esporádica no Brasil permitindo maior celeridade e confiabilidade no processo diagnóstico para elaborar um plano terapêutico mais adequado para a patologia e aporta informação com maior segurança ao paciente e seus familiares.
REFERÊNCIAS
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):13644.
- Atkinson CJ, Zhang K, Munn AL, Wiegmans A, Wei MQ. Prion protein scrapie and the normal cellular prion protein. Prion. 2016;10(1):63-82.
- M. Pocchiari, M. Puopolo, E. A. Croes, H. Budka, E. Gelpi, S. Collins, V. Lewis, T. Sutcliffe, A. Guilivi, N. Delasnerie-Laupretre, J.-P. Brandel, A. Alperovitch, I. Zerr, S. Poser, H. A. Kretzschmar, A. Ladogana, I. Rietvald, E. Mitrova, P. Martinez-Martin, J. de Pedro-Cuesta, M. Glatzel, A. Aguzzi, S. Cooper, J. Mackenzie, C. M. van Duijn, R. G. Will, Predictors of survival in sporadic Creutzfeldt–Jakob disease and other human transmissible spongiform encephalopathies, Brain, Volume 127, Issue 10, October 2004, Pages 2348–2359.
- Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005; 64: 1586–91
- Hermann, Peter, Brian Appleby, Jean-Philippe Brandel, Byron Caughey, Steven Collins, Michael D Geschwind, Alison Green, Stephane Haïk, Gabor G Kovacs, Anna Ladogana, Franc Llorens, Simon Mead, Noriyuki Nishida, Suvankar Pal, Piero Parchi, Maurizio Pocchiari, Katsuya Satoh, Gianluigi Zanusso, and Inga Zerr. “Biomarkers and Diagnostic Guidelines for Sporadic Creutzfeldt-Jakob Disease.” Lancet Neurology 20.3 (2021): 235-46. Web.
- CDC. Centers of Disease Control and Prevention. Creutzfeldt-Jakob Disease, Classic (CJD). EUA, 2021. Disponível em: https://www.cdc.gov/prions/cjd/ index.html
- McKenzie N, Piconi G, Culeux A, Hammarin AL, Stergiou C, Tzartos S, Versleijen AAM, van de Geer J, Cras P, Cardone F, Ladogana A, Mannana A, Rossi M, Bongianni M, Perra D, Regelsberger G, Klotz S, Hornemann S, Aguzzi A, Schmitz M, Andrews M, Burns K, Haïk S, Ruiz-García R, Verner-Carlsson J, Tzartos J, Verbeek MM, De Vil B, Poleggi A, Parchi P, Zanusso G, Gelpi E, Frontzek K, Reimann R, Hermann P, Zerr I, Pal S, Green A. Concordância da conversão induzida por tremores em tempo real do líquido cefalorraquidiano na Rede Europeia de Vigilância da Doença de Creutzfeldt-Jakob. Eur J Neurol. 2022 Agosto; 29(8):2431-2438. DOI: 10.1111/ene.15387. EPub 2022 25 de maio. PMID: 35524506; PMCID: PMC9543645.
¹Residente Neurologia SESPB
²Neurologista Preceptor Residência Neurologia SESPB