EVALUATION OF GENETIC VARIANTS IN THE JAK2 GENE IN PATIENTS WITH POLYCYTHEMIA VERA: AN INTEGRATIVE REVIEW
REGISTRO DOI:10.5281/zenodo.10070587
Patrícia Verena De Almeida1
Lucas Glouver Brandão De Moura2
Prof. Msc Gabriel De Oliveira Rezende3
RESUMO
A Policitemia vera é uma das três malignidades mieloides derivadas de células-tronco comumente conhecidas como neoplasias mieloproliferativas, caracterizada pelo acúmulo exacerbado de um ou mais elementos da linhagem eritróide, geralmente acompanhada de leucocitose e trombocitose. Embora a morfologia, análise clínica e laboratorial tenham um papel importante na descrição dessas neoplasias, a sinalização de JAK2 desregulada tornou-se objetivo de estudo central e a análise genômica está fornecendo uma plataforma para melhor compreensão dessas doenças. A descoberta de variante somática JAK2V617F na patogênese das neoplasias mieloproliferativas revolucionou o diagnóstico dessas doenças. Quase todos os pacientes com PV apresentam uma variante no gene JAK2. Essas variantes ativam de forma constitutiva a via de sinalização JAK-STAT, uma via intracelular envolvida na hematopoiese, resposta imune e ativação de outras vias de sinalização intracelular. Tendo em vista o conhecimento dessas variantes em estudos anteriores, a análise do cenário atual das variantes em JAK2, podem estar mudando ao longo do tempo, seja por fatores genéticos, ambientais ou de seleção clonal. O objetivo desse estudo é identificar as variantes genéticas no gene JAK2, explorando suas implicações clínicas e seu potencial impacto na Policitemia vera. Com base na literatura científica publicadas no período de 2013 a 2023, de acordo com os critérios de metodologia definidos. Os estudos avaliados permitiram o conhecimento do potencial de outras variantes presentes nos éxons 13 e 25, como possíveis indutores de instabilidade genômica pelo ganho de função na via JAK-STAT. Logo, entende-se a importância de mais estudos acerca dos mecanismos funcionais e patogênicos dessas variantes para melhor entendimento e abordagem terapêutica.
Palavras–chaves: Janus Quinase 2. Variantes genéticas. Neoplasias mieloproliferativas.
ABSTRACT
Polycythemia vera is one of three stem cell-derived myeloid malignancies commonly known as myeloproliferative neoplasms, characterized by the exacerbated accumulation of one or more elements of the erythroid lineage, generally accompanied by leukocytosis and thrombocytosis. Although morphology, clinical and laboratory analysis play an important role in describing these neoplasms, dysregulated JAK2 signaling has become a central objective of study and genomic analysis is providing a platform for better understanding these diseases. The discovery of the JAK2V617F somatic variant in the pathogenesis of MPNs revolutionized the diagnosis of these diseases. Almost all patients with PV have a variant in the JAK2 gene. These variants constitutively activate the JAK-STAT signaling pathway, an intracellular pathway involved in hematopoiesis, immune response and activation of other intracellular signaling pathways. Given the knowledge of these variants in previous studies, the analysis of the current scenario of variants in JAK2 may be changing over time, whether due to genetic, environmental or clonal selection factors. The objective of this study is to identify genetic variants in the JAK2 gene, exploring their clinical implications and their potential impact on Polycythemia vera. Based on scientific literature published from 2013 to 2023, in accordance with the defined methodology criteria. The studies evaluated allowed us to understand the potential of other variants present in exons 13 and 25, as possible inducers of genomic instability due to gain of function in the JAK-STAT pathway. Therefore, we understand the importance of further studies on the functional and pathogenic mechanisms of these variants for a better understanding and therapeutic approach.
Keywords: Janus Kinase 2.Genetic mutations.Myeloproliferative neoplasms.
1. Introdução
As neoplasias mieloproliferativas (MPNs) são doenças hematológicas clonais de células-tronco, conceituadas pela primeira vez em 1951 por William Dameshek e caracterizadas pelo excesso descontrolado de células mieloides maduras na medula óssea e, que geralmente levam ao aumento de contagem de células sanguíneas na circulação periférica (TEFFERI, 2016).
De acordo com a 5ª edição da Classificação da Organização Mundial da Saúde de Tumores Hematolinfóides (2022), o termo neoplasias mieloproliferativas é reservado as patologias sendo PV, TE e MF, as mais frequentes, embora inclua outras entidades clinicopatológicas relacionadas, como leucemia mieloide crônica (LMC), leucemia neutrofílica crônica (CNL), leucemia eosinofílica crônica (CEL), leucemia mielomonocítica juvenil (JMML) e neoplasia mieloproliferativa, não especificada de outra forma (MPN-NOS). A ênfase na distinção entre policitemia vera (PV), trombocitemia essencial (TE) e mielofibrose primária (MF) ainda é considerada, as quais são definidas pela ausência do rearranjo BCR/ABL1 e se baseia na integração de achados de sangue periférico, dados moleculares (mediadas por variantes drivers JAK2, CALR ou MPL) e achados de avaliação morfológica da medula óssea (KHOURY et al., 2022).
Essas neoplasias apresentam-se, em indivíduos entre a sexta e a sétima década de vida. PV apresenta incidência de 0,5 – 4,0 casos a cada 100.000 habitantes/ano, e é definida pela produção excessiva de hemácias e liberação de citocinas pró-inflamatórias. A sobrevida em policitemia vera é inferior à da TE, mas superior à da PMF, com medianas estimadas de 14, 20 e 6 anos, respectivamente (TEFFERI; VANNUCCHI; BARBUI, 2018).
Duas categorias de risco são consideradas: alta (idade > 60 anos ou história de trombose presente) e baixa (ausência de ambos os fatores de risco). A trombose de veia porta (TVP) ou hepática é uma complicação grave e a síndrome de Budd-Chiari (BDS) pode ser a característica clínica de apresentação tanto de PV quanto de TE com alta prevalência de variante JAK2V617F (SMALBERG et al., 2012).
As características clínicas da PV incluem grau leve a moderado de esplenomegalia e sintomas constitucionais, incluindo fadiga e prurido, sintomas de hiperviscosidade, leucocitose, trombocitose, sintomas microvasculares (ex. dor torácica, eritromelalgia, parestesia), trombose, complicações hemorrágicas e risco de transformação leucêmica ou progressão fibrótica (MCMULLIN et al., 2019).
Pacientes com PV geralmente apresentam uma variante no gene JAK2 (Janus quinase 2)(HGNC ID: 6192). Este, está localizado no cromossomo 9p24.1 e possui 142.939 pares de bases (pb) nos quais estão localizados a região promotora, possuindo 25 éxons, 25 íntrons e a região terminadora (PAES et al., 2022), cerca de 96% e 3% exibindo alterações genéticas de ativação somática no éxon 14 (JAK2V617F) e éxon 12 de JAK2, respectivamente (TEFFERI; BARBUI, 2020b).
Essas variantes ativam de forma constitutiva a via de sinalização JAK-STAT, uma via intracelular envolvida na hematopoiese, resposta imune e ativação de outras vias de sinalização intracelular. Em PV, há um aumento da fosforilação das proteínas STAT5, STAT3 e STAT5, além disso, esta última demonstrou ser essencial para o desenvolvimento de PV com normalização das contagens sanguíneas e tamanho do baço ocorrendo em um modelo de camundongo knockout STAT5 (O’SULLIVAN; HARRISON, 2017).
Desde a descoberta de JAK2V617F (dbSNP ID: rs77375493), caracterizada por uma substituição de base do tipo transversal no nucleotídeo 1849 (1849G > T) do éxon 14 do gene JAK2, causando a substituição de valina para fenilalanina no códon 617 (V617F). A variante apresenta frequência maior que 95% em indivíduos com PV, e frequência de 55% a 65% em indivíduos com TE e MF, respectivamente sua associação se tornou base molecular para o diagnóstico em NMP’s (TORRES et al., 2022).
A ativação das variantes em JAK2 podem surgir de translocações cromossômicas ou variantes/deleções/inserções pontuais (TEFFERI, 2016). Contudo, mesmo que JAK2V617F seja suficiente para causar proliferação de precursores eritropoéticos, a heterogeneidade da doença sugere a manifestação de fenômenos adicionais que contribuem para a leucemogênese.
Tendo em vista o conhecimento dessas variantes em estudos anteriores, e análise do cenário atual das variantes em JAK2, podemestar mudando ao longo do tempo, seja por fatores genéticos, ambientais ou de seleção clonal. Um estudo retrospectivo sistematizado pode fornecer informações valiosas sobre a prevalência, distribuição e características verificadas no perfil das variantes genéticas no gene JAK2 em amostras de pacientes diagnosticados com Policitemia Vera, e como essas variantes genéticas podem estar associadas à patogênese, ao curso clínico e à resposta ao tratamento da doença.
2. Material e Métodos
Para a realização deste artigo de revisão narrativa da literatura, procedeu-se a uma pesquisa por meio de bases de dados como: PubMed, Science direct e Portal Regional da BVS e Google Acadêmico utilizando os operadores booleanos (OR;AND) e os seguintes descritores em português, definidas pelo DeCS (Descritores em Ciências da Saúde): (transtornos mieloproliferativos) OR (Policitemia Vera) AND (polimorfismo de nucleotídeo único) OR (taxa de variante) OR (variante) AND (Janus Quinase 2) bem como os descritores MeSH (Medical Subject Headings Terms) em inglês: (((((“Polycythemia Vera”[Mesh]) OR “Myeloproliferative Disorders”[Mesh]) AND “Polymorphism, Single Nucleotide”[Mesh]) OR “Mutation”[Mesh]) OR “Mutation Rate”[Mesh]) AND “Janus Kinase 2″[Mesh].
Foram selecionados artigos em língua portuguesa, inglesa ou espanhol publicados no período de 2013-2023, identificados como relatos de caso, estudos de coorte, estudos de Caso-Controle ou Ensaios Clínicos, revisões e meta-análises sendo assim excluídos artigos duplicados, textos incompletos, e aqueles que fugiram da temática abordada neste estudo ou não apresentavam uma maneira de acessá-los foram descartados.
Por fim, 57 dos 480 artigos foram selecionados para a produção desta revisão. Vale ressaltar que livros e documentos importantes do Ministério da Saúde foram incorporados ao estudo.
A figura a seguir ilustra a metodologia de forma mais didática:
Figura 1. Esquematização da busca e da seleção das referências bibliográficas.
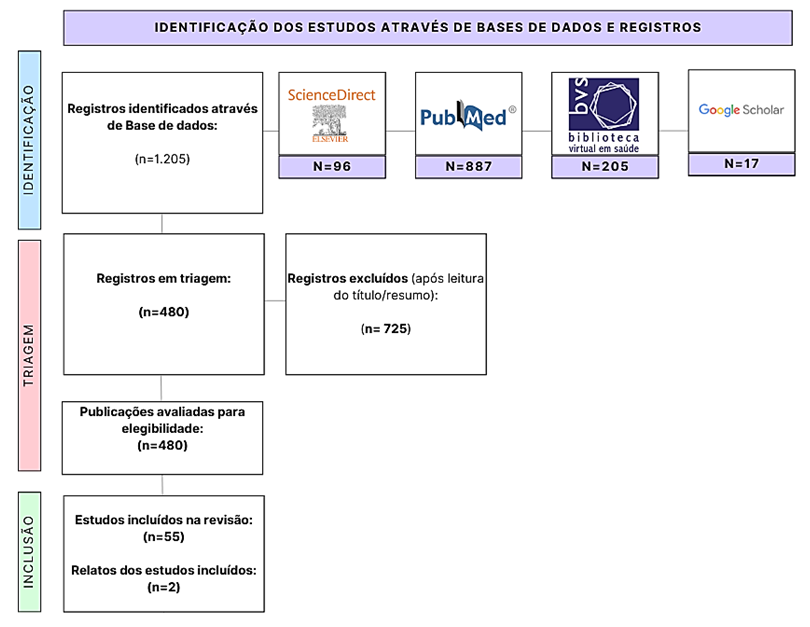
Fonte: Autoria própria, 2023.
3. Referencial teórico
3.1. Base genética da Policitemia Vera
Em 1976, descobriu-se a natureza celular da Policitemia Vera e a evidência de sua origem clonal ao analisar a inativação do cromossomo X no sangue periférico de duas mulheres (ADAMSON et al., 2009). Mais tarde, essa descoberta foi confirmada pela identificação de variantes genéticas somáticas como marcadores clonais (VAINCHENKER; KRALOVICS, 2017). Com o avanço do sequenciamento de próxima geração (NGS), o conhecimento sobre as variantes envolvidas nas NMPs progrediu significativamente, proporcionando uma compreensão mais aprofundada da base genética dessas doenças, com o objetivo de melhorar e personalizar as previsões prognósticas para cada paciente (LUQUE PAZ; KRALOVICS; SKODA, 2023).
A presença de duas ou mais variantes somáticas teve um impacto significativo na redução da sobrevida global e no aumento do risco de desenvolver leucemia mieloide aguda (LUNDBERG et al., 2014). Uma variante comum encontrada na maioria dos pacientes com neoplasias mieloproliferativas é a JAK2V617F (BAXTER et al., 2005).
Embora tenha sido comprovado que as variantes em JAK2 desempenham um papel importante nas características da doença, existem evidências de que eventos mutacionais e clonais ocorrem antes da aquisição da variante JAK2V617F, isso sugere que a evolução da doença envolve múltiplos estágios e fatores genéticos (KRALOVICS et al., 2006).
Contudo, Luque Paz et al. (2023) explicam que as “variantes passageiras” são alterações na sequência genética sem impacto imediato na função. Elas são úteis como marcadores para identificar subclones em neoplasias mieloproliferativas (NMPs). No entanto, determinar se uma variante afeta a função do gene é desafiador, pois muitas não foram analisadas detalhadamente. Isso limita a compreensão dessas patologias (LUQUE PAZ; KRALOVICS; SKODA, 2023).
Figura 2. Reconstruindo a linha do tempo da evolução clonal em neoplasias mieloproliferativas.
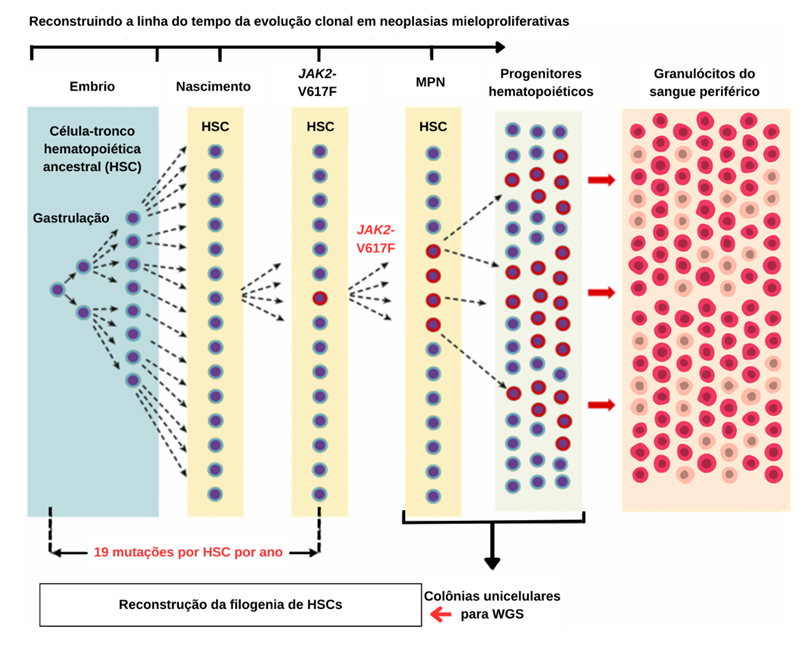
Fonte: Adaptado de Luque Paz et al. 2023.
Na Figura 2, vemos a evolução das NMPs devido à variante JAK2V617F desde a embriogênese, com células-tronco hematopoiéticas. As variantes nas células-filhas servem como marcadores de linhagem. Novas variantes acumulam a cada divisão celular, formando uma árvore filogenética com base nas sequências genéticas. Para estudar isso, foi feito um estudo em células da medula óssea após o diagnóstico, cultivaram HSCs ou células progenitoras em poços individuais e posteriormente sequenciado o DNA de cada colônia por meio de análise genômica completa (WGS) e assim construída a árvore filogenética a partir do ancestral comum das HSCs durante a gastrulação (LEE-SIX et al., 2018).
Para estimar quando a variante JAK2V617F foi adquirida, considera-se uma taxa constante de 19 variantes por HSC por ano. Importante destacar que essa estimativa não é influenciada pelo aumento na taxa de divisão celular das HSCs mutantes de JAK2V617F, pois apenas as alterações genéticas que ocorreram antes da aquisição dessa variante foram utilizadas para calcular essa estimativa. Esse método ajuda a compreender a trajetória da doença ao longo do tempo e a sua origem a partir de um ancestral comum das células-tronco hematopoiéticas (VAN EGEREN et al., 2021; WILLIAMS et al., 2022)
Desenvolvimentos recentes em técnicas de sequenciamento altamente sensíveis, com o uso de códigos de barras específicos de cadeias, possibilitaram a detecção de variantes em nível de molécula única. No entanto, a perda da informação sobre o tipo de célula, é um desafio a menos que classificação das células seja antes do sequenciamento. Isso limita nossa compreensão dos padrões de variantes somáticas entre diferentes tipos de células e seu impacto nos destinos e fenótipos celulares. Uma abordagem alternativa envolve a detecção de variantes somáticas diretamente em leituras de sequenciamento de ensaios unicelulares de alto rendimento, como sequenciamento de RNA unicelular (scRNA-seq) e sequenciamento de cromatina acessível por transposase (scATAC-seq)(RANZONI et al., 2021; VAN GALEN et al., 2019) aproveitando o alto rendimento desses ensaios para mapear a linhagem celular em programas de transcrição ou regulação sem a necessidade de protocolos complexos para perfilar o DNA e RNA da mesma célula (FAN et al., 2018; PETTI et al., 2019).
Um algoritmo chamado SComatic foi desenvolvido para detectar variantes somáticas em conjuntos de dados de perfil de célula única, sem a necessidade de dados de sequenciamento de DNA correspondente, permitindo a análise da mutagênese somática em grande escala, abrindo possibilidades para estudos em humanos e outros organismos (MUYAS et al., 2023).
3.2. O gene Janus Quinase 2
O gene Janus quinase 2 (HGNC ID: 6192) está localizado no cromossomo 9p24.1 e possui 142.939 pares de bases (pb) nos quais estão localizados a região promotora, 25 éxons, 25 íntrons e a região terminadora (PAES et al., 2022), cerca de 96% e 3% exibindo alterações genéticas de ativação somática no éxon 14 (JAK2V617F) e éxon 12 de JAK2, respectivamente (TEFFERI; BARBUI, 2020b).
Figura 3. A. Representação esquemática de JAK2 (NG_009904) e variantes genéticas definidas como critérios de diagnóstico em Policitemia Vera (em vermelho) A variante JAK2V617F caracterizada por uma substituição de base do tipo transversão no nucleotídeo 1849 (1849G > T) do éxon 14.B. Estrutura modular de JAK2, que se associa aos receptores de citocinas tipo II e à maioria dos receptores de citocinas tipo I. Os JAKs têm sete domínios homólogos (JH1-7), incluindo o domínio catalítico (JH1) e o domínio pseudoquinase cataliticamente inativo (JH2), que supostamente regula negativamente a atividade do domínio quinase. O domínio JH3-JH4 dos JAKs compartilha homologia com os domínios Src homology 2 (SH2). O domínio amino terminal (JH4-JH7) é conhecido como domínio FERM, que se envolve com receptores de citocinas.
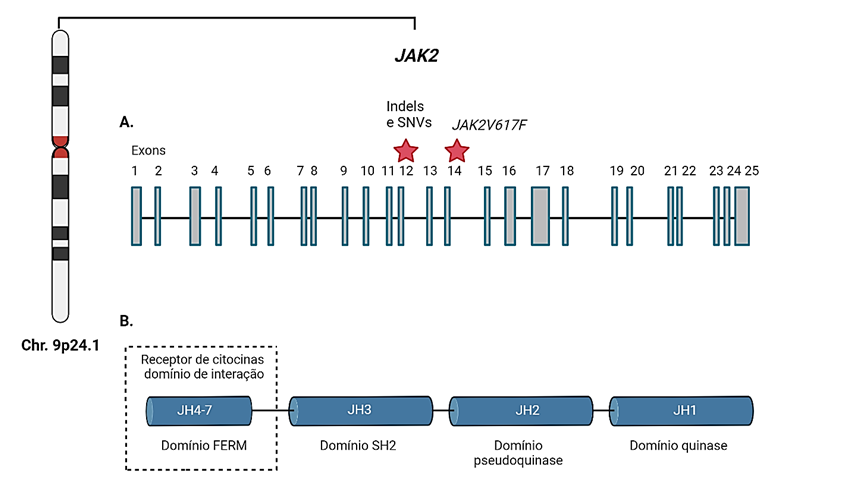
Fonte: Adaptado de Anelli et al; Paes et al. 2022.
A família JAK é composta por proteínas tirosina quinases não receptoras que desempenham um papel importante na regulação de sinais celulares quando as citocinas se conectam aos seus receptores. Essa família é composta por quatro membros principais: JAK1, JAK2, JAK3 e TYK2. Cada membro da família JAK possui sete domínios de homologia, chamados de domínios de homologia JAK (JH). O primeiro deles, JH1, também conhecido como domínio quinase, contém cerca de 250 resíduos de aminoácidos e codifica uma proteína quinase que desempenha um papel fundamental na fosforilação de substratos (TEFFERI; BARBUI, 2020).
O segundo domínio, JH2, é chamado de domínio PK e, embora seja estruturalmente semelhante ao domínio quinase, não possui ação quinase. Este atua principalmente regulando a atividade do domínio quinase. Além disso, há o domínio pseudoquinase, que interage com proteínas STAT, e o domínio PK, que pode inibir a atividade da tirosina quinase. Por fim, os domínios JH3, metade de JH4, JH5, JH6 e JH7 juntam-se para formar o domínio FERM. Estes diversos domínios desempenham papéis cruciais na sinalização celular e na regulação da atividade das proteínas JAK (FERRAO; LUPARDUS, 2017; HU et al., 2021).
As proteínas STAT são fatores de transcrição intracelular que desempenham um papel vital na regulação de processos como imunidade, proliferação celular e diferenciação. Existem sete tipos de STAT, ativados por diferentes combinações de citocinas, hormônios de crescimento e proteínas JAK. Para sua ativação, STATs requerem a formação de um complexo receptor que envolve citocinas específicas e proteínas JAK. Essa ativação direcionada desencadeia respostas celulares específicas, desempenhando um papel central na coordenação das funções celulares e na resposta a estímulos externos (BALDINI et al., 2021).
Curiosamente, foi demonstrado que as proteínas STAT são ativadas diferencialmente nos MPNs; o aumento da fosforilação de STAT5 e STAT3 ocorre em PV, mas o aumento de STAT3 e a redução da fosforilação de STAT5 em ET. Os pacientes com MF tiveram fosforilação reduzida de ambos. Demonstrou-se que o STAT5 é obrigatório para o desenvolvimento de PV, com normalização do hemograma e do tamanho do baço ocorrendo em um modelo de camundongo knockout para STAT5 (O’SULLIVAN; HARRISON, 2017).
3.3. Perspectivas atuais sobre os inibidores de JAK
Na área da hematologia, os inibidores de JAK têm se destacado principalmente no tratamento das neoplasias mieloproliferativas. O ruxolitinibe, aprovado pelo FDA dos EUA e pela EMA em 2011, foi o pioneiro nesse cenário como um inibidor de JAK1 e JAK2 (PASSAMONTI et al., 2017).
Apesar dos avanços, ainda faltam mais estudos sobre a especificidade na sinalização JAK-STAT, visando melhorar a eficácia clínica e reduzir os efeitos colaterais. Vários ensaios clínicos, concluídos e em andamento, estão explorando o uso desses inibidores nas neoplasias mieloproliferativas, tanto como agentes únicos quanto em combinação com inibidores de JAK (PETTIT et al., 2022).
Atualmente, os inibidores de JAK licenciados incluem tofacitinibe, ruxolitinibe, fedratinibe, upadacitinibe, peficitinibe e baricitinibe. No contexto da policitemia vera, o estudo RESPONSE mostrou que o ruxolitinibe superou a melhor terapia disponível, tornando-se uma opção importante para pacientes resistentes à hidroxicarbamida. O ruxolitinibe também obteve aprovação como tratamento de segunda linha na policitemia vera, o que pode reduzir complicações trombóticas a longo prazo (KILADJIAN et al., 2020; PASSAMONTI et al., 2017).
No entanto, é importante ressaltar que os inibidores de JAK podem ter limitações na neoplasia mieloproliferativa, como doses inadequadas e perda de resposta (MCLORNAN et al., 2021).
3.4. A via de sinalização JAK-STAT
Na ausência de citocinas, a proteína JAK permanece inativa, localizada próxima aos domínios intracelulares do receptor. Quando uma citocina se liga ao receptor, ocorre a fosforilação das proteínas JAK e dos domínios intracelulares do receptor, ativando-os. Isso leva ao recrutamento de proteínas STAT, que se agrupam (dimerizam) e migram para o núcleo da célula. Esse processo inicia a transcrição de genes envolvidos na proliferação celular (TORRES et al., 2022).
Além da ativação da via JAK/STAT, ocorre uma interação com outras vias de sinalização intracelular, incluindo as vias Ras/Raf/MAPK e PI3K. A via PI3K, em particular, está conectada indiretamente às proteínas JAK1, que ativam a via NFkB. Essas vias de sinalização desempenham um papel importante na produção de citocinas, como CXCL12, IL-6, IL-8, IL-9, TNF-α e CCL3, e estão relacionadas com o crescimento de fatores presentes em perfis inflamatórios de indivíduos com neoplasias mieloproliferativas crônicas (CLARK; FLANAGAN; TELLIEZ, 2014).
Em diversos estados patológicos, a via JAK-STAT não funciona de maneira normal devido a variantes, tanto esporádicas quanto hereditárias, em elementos de sinalização diretamente envolvidos na via ou em moléculas reguladoras. Isso pode resultar em diferentes manifestações clínicas, dependendo das combinações defeituosas de ligantes, proteínas JAK e STAT, sequências de DNA ou proteínas reguladoras em comparação com a situação fisiológica. Como resultado, os distúrbios nessa via podem contribuir para doenças reumatológicas ou neoplasias mieloproliferativas (BALDINI et al., 2021).
A variante JAK2V617F resulta na ativação constante da proteína JAK2, causando a perda da função inibitória do domínio pseudoquinase e ativando a sinalização intracelular através das proteínas STAT, bem como das vias de proteína quinase ativada por mitógeno e fosfoinositídeo 3-quinase. Isso leva a uma proliferação excessiva de células mieloides e diferenciação. Variantes incomuns, como as do éxon 12 de JAK2, também podem ativar a sinalização JAK-STAT de forma constitutiva, contribuindo para estados patológicos (NANGALIA; GREEN, 2017; SCOTT et al., 2007). Estes foram descritos abaixo na Tabela 1.
Tabela 1. Resumo das mutações canônicas e atípicas de JAK2 em PV.
Referência Periódico Localização/Variante/Tipo Resultados encontrados Significado Clínico (PATEL et al., 2019) Blood Variante Exon 13 de JAK2/JAK2ex13InDel/ inserção/deleção Está associada à eosinofilia e eritrocitose, possivelmente representando uma nova entidade clínica. JAK2ex13InDel leva à ativação constitutiva e promove sinalização através de receptores baseados em cadeia comum β na ausência de ligante. Tanto os pacientes com PV como com CEL apresentam alto risco de trombose, e a citorredução concomitante de glóbulos vermelhos, neutrófilos e eosinófilos pode ser necessária para a prevenção de eventos tromboembólicos. (WU et al., 2014) Journal of Hematology & Oncology Variantes Exon12 de JAK2:
M532V/E543G,N533D,M535I/H538Y/K549I, E543G e D544NPacientes chineses tiveram variantes no exon 12 de JAK2 muito mais difundidas (13%) do que nos ocidentais e outros asiáticos orientais. As variantes do Exon 12 do JAK2 foram prevalentes (13%) e variáveis nos pacientes chineses. Em comparação com pacientes com PV com variantes JAK2V617F, os pacientes com PV com variantes no exon 12 de JAK2 tiveram um início mediano mais precoce da doença (ANBINSELVAM et al., 2020) Blood Cells, Molecules, and Diseases Variantes Exon 12 deJAK2:
g.84941C>T(rs569230287) / missense e g.84993_84995delinsTT/ indelg.84941C > T (rs569230287) é uma variante que leva à substituição da prolina pela leucina na posição 521 (P521L). Ao passo que a variante g.84993_84995 delinsTTT leva à substituição de lisina por leucina na posição 539 (K539L) e foi associada à ativação constitutiva de JAK2. g.84993_84995 delinsTTT- O paciente portador dessa variante indel apresentava nível sérico normal de eritropoetina (2,4 mUI/mL), mas a hemoglobina (20,9 g/dL), o hematócrito (62,5%) e a contagem de hemácias (7,21 M/uL) estavam elevados. (PITA et al., 2018) Journal of Clinical Pathology Variante Exon 12 de JAK2:
c.1605G>T(p.Met535Ile) e c.1612C>T (p.His538Tyr)Uma ativação aumentada de STAT1 pode causar megacariopoiese superior e, portanto, possivelmente ser um fator que contribui para a compreensão da pequena parcela de pacientes com PV com variante no exon 12 que se comportam de maneira diferente. Por outro lado, a ausência de trombocitose na maioria dos pacientes pode estar relacionada a mecanismos independentes do STAT1. A base para o fenótipo diferente do paciente não é conhecida. (OLIVEIRA E COSTA et al., 2023) Hematology, Transfusion and Cell Therapy Exon 25 de JAK2/JAK2:c.3323A>G(p.Asn1108Ser)/ variante germinativa Classificada como provavelmente patogênica na análise in-silico, pois poderia representar um ganho de função na via JAK-STAT. O significado e função desta variante ainda permanecem incertos. (MOLITERNO; KAIZER; REEVES, 2023)
(REGIMBEAU et al., 2022)Blood Exon 14 de JAK2:
JAK2V617F/somática (ganho de função)TERNO; KAIZER; REEVES, 2023)
(REGIMBEAU et al., 2022)A variante V617F tem efeitos ativadores na atividade de sinalização de JAK2, na sensibilidade às citocinas e na sobrevivência dependente de citocinas nas linhas celulares. Várias alterações de sinalização parecem mediar os efeitos celulares de JAK2V617F. O aumento da fosforilação de STAT5 foi demonstrado. Cerca de 30% dos pacientes apresentaram um nível de hemoglobina significativamente mais elevado, aumento da incidência de prurido, eritropoiese e mielopoiese estimuladas, maior prevalência de esplenomegalia e aumento de células progenitoras no sangue periférico.
3.5. Variantes canônicas JAK2
A variante JAK2V617F (c.1849G> T, p. Val617Phe) é uma variante somática de ganho de função. Sua presença é um importante critério diagnóstico para NMP negativo para cromossomo Filadélfia (CREE, 2022). O mutante JAK2V617F ativa as vias de sinalização JAK‐STAT, PI3K/Akt e ERK1/2 MAPK, levando à expansão anormal das células mieloides. O mutante JAK2V617F induz a trombogênese por diferentes mecanismos: maior hematócrito com alta viscosidade sanguínea, interações plaquetas-endotélio aumentadas, agregados plaquetas-leucócitos e aumento da produção de citocinas pró-inflamatórias (REGIMBEAU et al., 2022). Eventos trombóticos foram observados em 30,2% dos pacientes com a variante JAK2V617F, em comparação com 9,2% dos pacientes que não a apresentavam (FOREST et al., 2020). A frequência de variantes na JAK2 aponta a variante V617F como a principal presente em 97% dos casos, porém, apesar disso, a presença da variante V617F não impede a existência de outras variantes no éxon 14 ou de outros éxons, sendo estes cerca de 3% dos casos (TEFFERI; BARBUI, 2023).
3.6. Variantes não canônicas JAK2
3.6.1. Variantes do Éxon 12
Segundo as diretrizes da OMS de 2022, um dos principais critérios de diagnóstico para PV, são variantes NM_004972: JAK2 p.V617F ou JAK2 no exon 12, e ao contrário da substituição V617F, variantes no exon 12 de JAK2 são encontradas exclusivamente na PV (CREE, 2022; TONDEUR et al., 2021).
Este éxon codifica aminoácidos de 505 a 547, suas variantes identificadas vão dos códons de 533 a 547, pelo menos 27 variantes do éxon 12 clinicamente avaliadas foram identificadas até o momento, incluindo substituição de aminoácidos, deleções e duplicações (GONG et al., 2013; MADDALI et al., 2020). Estas podem constituir cerca de 2-5% dos casos negativos para JAK2V617F. Portanto, os pacientes devem ser analisados para outras variantes de interesse, como a K539L, que leva à substituição de lisina por leucina na posição 539 do exon 12, e está associada a ativação constitutiva da JAK2, e resultando em fenótipos variáveis(ANBINSELVAM et al., 2020; MADDALI et al., 2020).
Na distribuição das linhagens sanguíneas, as variantes do éxon 12 podem ser rastreadas em porcentagens variáveis de granulócitos, monócitos e plaquetas, porém raramente linfócitos (FURTADO et al., 2013). Eritrocitose isolada e eritropoetina suprimida são características clínicas frequentes em pacientes com variantes no éxon 12, ademais, geralmente possuem medula óssea com morfologia inespecífica e proliferação eritróide isolada (GONG et al., 2013).
As variantes mais frequentes do exon 12 envolvem uma deleção in-frame de seis nucleotídeos nos códons 542 e 543 (N542_E543del), que representa aproximadamente 40% dos pacientes com PV negativos para a variante V617F (GONG et al., 2013; REGIMBEAU et al., 2022).
3.6.2. Variantes do Éxon 13
Algumas variantes mais raras podem causar a PV, dentre elas estão as do éxon 13 do gene JAK2, sendo R564L, R564Q, V567A, G571S, G571R, L579F, H587N, S591L e F557L as mais descritas (REGIMBEAU et al., 2022). Um estudo recente identificou por meio do sequenciamento direto uma variante silenciosa no éxon 13 no códon 570, além disso no mesmo estudo, dos cinco pacientes portadores da variante missense p.G571S no éxon 13, um apresentou trombocitose e embolia pulmonar. Logo, entende-se que a variante p.G571S pode induzir uma mudança conformacional, tornando o Y570 inacessível e impedindo sua fosforilação e produzindo assim, uma quinase JAK2 constitutivamente ativa (ALGHASHAM et al., 2016).
3.6.3. Variante do Éxon 25: O caso clínico
Um recente estudo realizado no Brasil relatou um caso clínico onde o paciente negativo para variante JAK2V617F e variantes do éxon 12 foi positivo para uma variante no éxon 25 do gene JAK2 (JAK2:c.3323A>G (p.Asn1108Ser)), este foi possível por meio de sequenciamento por meio da técnica de próxima geração (NGS) Custom Ampliseq ION S5 (OLIVEIRA E COSTA et al., 2023).
Oliveira E Costa et al. (2023), classificou esta variante como provável patogênica em sua análise in-silico, pois poderia representar um ganho de função na via JAK-STAT. Além disso, a variante N1108S, foi mais comum no grupo que desenvolveu LMA. Estes dados sugerem a potencial indução de instabilidade genômica dessa variante, favorecendo a mutagênese e a leucemogênese (FERNÁNDEZ et al., 2015; OLIVEIRA E COSTA et al., 2023).
4. CONCLUSÃO
Variantes genéticas da JAK2 estão ativamente envolvidas no processo de patogênese da PV, a literatura já vem demonstrando sua importância, principalmente nos éxons 14 (JAK2V617F) e éxon 12 para a doença. Os estudos avaliados permitiram o conhecimento do potencial de outras variantes presentes nos éxons 13 e 25, como possíveis indutores de instabilidade genômica pelo ganho de função na via JAK-STAT. Logo, entende-se a importância de mais estudos acerca dos mecanismos funcionais e patogênicos dessas variantes para melhor entendimento e abordagem terapêutica.
REFERÊNCIAS
ADAMSON, J. W. et al. Polycythemia Vera: Stem-Cell and Probable Clonal Origin of the Disease. http://dx.doi.org/10.1056/NEJM197610212951702, v. 295, n. 17, p. 913–916, 21 nov. 2009.
ALGHASHAM, N. et al. Detection of mutations in JAK2 exons 12-15 by Sanger sequencing. International Journal of Laboratory Hematology, v. 38, n. 1, p. 34–41, 1 fev. 2016.
ANBINSELVAM, A. et al. Mutation profile of JAK2, EPOR and CALR genes in polycythemia patients. Blood Cells, Molecules, and Diseases, v. 82, p. 102414, 1 maio 2020.
BALDINI, C. et al. The JAK–STAT pathway: an emerging target for cardiovascular disease in rheumatoid arthritis and myeloproliferative neoplasms. European Heart Journal, v. 42, n. 42, p. 4389, 11 nov. 2021.
BAXTER, E. J. et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet, v. 365, n. 9464, p. 1054–1061, 19 mar. 2005.
CLARK, J. D.; FLANAGAN, M. E.; TELLIEZ, J. B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. Journal of medicinal chemistry, v. 57, n. 12, p. 5023–5038, 26 jun. 2014.
CREE, I. A. The WHO Classification of Haematolymphoid Tumours. LeukemiaSpringer Nature, , 1 jul. 2022.
FAN, J. et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Research, v. 28, n. 8, p. 1217–1227, 1 ago. 2018.
FERNÁNDEZ, C. et al. Molecular charcterization of myeloproliferative neoplasms evolved to acute myeloid leukemia. Clinical Lymphoma Myeloma and Leukemia, v. 15, p. S16–S17, set. 2015.
FERRAO, R.; LUPARDUS, P. J. The Janus Kinase (JAK) FERM and SH2 domains: Bringing specificity to JAK-receptor interactions. Frontiers in Endocrinology, v. 8, n. APR, p. 255596, 18 abr. 2017.
FOREST, C. et al. Jak2 mutation expands the thrombophilic panel in children. Journal of Thrombosis and Haemostasis, v. 18, n. 10, p. 2636–2639, 1 out. 2020.
FURTADO, L. V. et al. A Multiplexed fragment analysis-based assay for detection of JAK2 exon 12 mutations. Journal of Molecular Diagnostics, v. 15, n. 5, p. 592–599, set. 2013.
GONG, J. Z. et al. Laboratory practice guidelines for detecting and reporting JAK2 and MPL mutations in myeloproliferative neoplasms: A report of the association for molecular pathology. Journal of Molecular Diagnostics, v. 15, n. 6, p. 733–744, nov. 2013.
HU, X. et al. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduction and Targeted Therapy 2021 6:1, v. 6, n. 1, p. 1–33, 26 nov. 2021.
KHOURY, J. D. et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022 36:7, v. 36, n. 7, p. 1703–1719, 22 jun. 2022.
KILADJIAN, J. J. et al. Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. The Lancet. Haematology, v. 7, n. 3, p. e226–e237, 1 mar. 2020.
KRALOVICS, R. et al. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood, v. 108, n. 4, p. 1377–1380, 15 ago. 2006.
LEE-SIX, H. et al. Population dynamics of normal human blood inferred from somatic mutations. Nature, v. 561, n. 7724, p. 473, 27 set. 2018.
LUNDBERG, P. et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood, v. 123, n. 14, p. 2220–2228, 3 abr. 2014.
LUQUE PAZ, D.; KRALOVICS, R.; SKODA, R. C. Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood, v. 141, n. 16, p. 1909–1921, 20 abr. 2023.
MADDALI, M. et al. JAK2 exon 12 mutations in cases with JAK2V617F-negative polycythemia vera and primary myelofibrosis. Annals of Hematology, v. 99, n. 5, p. 983–989, 1 maio 2020.
MCLORNAN, D. P. et al. Current and future status of JAK inhibitors. Lancet (London, England), v. 398, n. 10302, p. 803–816, 28 ago. 2021.
MCMULLIN, M. F. et al. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology Guideline. British journal of haematology, v. 184, n. 2, p. 176–191, 1 jan. 2019.
MOLITERNO, A. R.; KAIZER, H.; REEVES, B. N. JAK2 V617F allele burden in polycythemia vera: burden of proof. Blood, v. 141, n. 16, p. 1934–1942, 20 abr. 2023.
MUYAS, F. et al. De novo detection of somatic mutations in high-throughput single-cell profiling data sets. Nature Biotechnology 2023, p. 1–10, 6 jul. 2023.
NANGALIA, J.; GREEN, A. R. Myeloproliferative neoplasms: from origins to outcomes. Blood, v. 130, n. 23, p. 2475–2483, 7 dez. 2017.
OLIVEIRA E COSTA, A. et al. Polycythemia and JAK2 variant N1108S: cause-and-effect or coincidence? Hematology, Transfusion and Cell Therapy, 21 fev. 2023.
O’SULLIVAN, J. M.; HARRISON, C. N. JAK-STAT signaling in the therapeutic landscape of myeloproliferative neoplasms. Molecular and Cellular Endocrinology, v. 451, p. 71–79, 15 ago. 2017a.
O’SULLIVAN, J. M.; HARRISON, C. N. JAK-STAT signaling in the therapeutic landscape of myeloproliferative neoplasms. Molecular and cellular endocrinology, v. 451, p. 71–79, 15 ago. 2017b.
PAES, J. et al. The Contribution of JAK2 46/1 Haplotype in the Predisposition to Myeloproliferative Neoplasms. International Journal of Molecular Sciences 2022, Vol. 23, Page 12582, v. 23, n. 20, p. 12582, 20 out. 2022.
PASSAMONTI, F. et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. The Lancet. Oncology, v. 18, n. 1, p. 88–99, 1 jan. 2017.
PATEL, A. B. et al. JAK2 ex13InDel drives oncogenic transformation and is associated with chronic eosinophilic leukemia and polycythemia vera. Blood, v. 134, n. 26, p. 2388–2398, 26 dez. 2019.
PETTI, A. A. et al. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nature Communications 2019 10:1, v. 10, n. 1, p. 1–16, 14 ago. 2019.
PETTIT, K. et al. Management of Myeloproliferative Neoplasms in the Molecular Era: From Research to Practice. American Society of Clinical Oncology educational book. American Society of Clinical Oncology. Annual Meeting, v. 42, n. 42, p. 595–613, jul. 2022.
PITA, A. S. A. et al. Atypical haematological presentation in a case of polycythaemia vera with a new variant mutation detected in exon 12: c.1605G>T (p.Met535Ile). Journal of Clinical Pathology, v. 71, n. 2, p. 180–184, 1 fev. 2018.
RANZONI, A. M. et al. Integrative Single-Cell RNA-Seq and ATAC-Seq Analysis of Human Developmental Hematopoiesis. Cell stem cell, v. 28, n. 3, p. 472- 487.e7, 4 mar. 2021.
REGIMBEAU, M. et al. Genetic Background of Polycythemia Vera. Genes, v. 13, n. 4, 2 abr. 2022.
SCOTT, L. M. et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. The New England journal of medicine, v. 356, n. 5, p. 459–468, fev. 2007.
SMALBERG, J. H. et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood, v. 120, n. 25, p. 4921–4928, 13 dez. 2012.
TEFFERI, A. Myeloproliferative neoplasms: A decade of discoveries and treatment advances. American Journal of Hematology, v. 91, n. 1, p. 50–58, 1 jan. 2016a.
TEFFERI, A. Somatic JAK2 mutations and their tumor phenotypes. Blood, v. 128, n. 6, p. 748–749, 11 ago. 2016b.
TEFFERI, A.; BARBUI, T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. American journal of hematology, v. 95, n. 12, p. 1599–1613, dez. 2020a.
TEFFERI, A.; BARBUI, T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. American Journal of Hematology, v. 95, n. 12, p. 1599–1613, 1 dez. 2020b.
TEFFERI, A.; BARBUI, T. Polycythemia vera: 2024 update on diagnosis, risk-stratification, and management. American Journal of Hematology, v. 98, n. 9, p. 1465–1487, 1 set. 2023.
TEFFERI, A.; VANNUCCHI, A. M.; BARBUI, T. Polycythemia vera treatment algorithm 2018. Blood Cancer Journal 2018 8:1, v. 8, n. 1, p. 1–7, 10 jan. 2018.
TONDEUR, S. et al. Long-term follow-up of JAK2 exon 12 polycythemia vera: a French Intergroup of Myeloproliferative Neoplasms (FIM) study. Leukemia, v. 35, n. 3, p. 871–875, mar. 2021.
TORRES, D. G. et al. JAK2 Variant Signaling: Genetic, Hematologic and Immune Implication in Chronic Myeloproliferative Neoplasms. Biomolecules 2022, Vol. 12, Page 291, v. 12, n. 2, p. 291, 11 fev. 2022a.
TORRES, D. G. et al. JAK2 Variant Signaling: Genetic, Hematologic and Immune Implication in Chronic Myeloproliferative Neoplasms. Biomolecules, v. 12, n. 2, 11 fev. 2022b.
VAINCHENKER, W.; KRALOVICS, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood, v. 129, n. 6, p. 667–679, 9 fev. 2017.
VAN EGEREN, D. et al. Reconstructing the Lineage Histories and Differentiation Trajectories of Individual Cancer Cells in Myeloproliferative Neoplasms. Cell stem cell, v. 28, n. 3, p. 514- 523.e9, 4 mar. 2021.
VAN GALEN, P. et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell, v. 176, n. 6, p. 1265- 1281.e24, 7 mar. 2019.
WILLIAMS, N. et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 2022 602:7895, v. 602, n. 7895, p. 162–168, 20 jan. 2022.
WU, Z. et al. The mutation profile of JAK2 and CALR in Chinese Han patients with Philadelphia chromosome-negative myeloproliferative neoplasms. Journal of Hematology & Oncology, v. 7, n. 1, p. 48–48, 15 jul. 2014.
1 Graduanda em Biomedicina. Faculdade Metropolitana de Manaus (FAMETRO). Av. Constantino Nery, 3000 – Chapada, Manaus – AM, CEP: 69050-000. E-mail: patverenamoni@gmail.com
2 Graduando em Biomedicina. Faculdade Metropolitana de Manaus (FAMETRO). Av. Constantino Nery, 3000 – Chapada, Manaus – AM, CEP: 69050-000. E-mail: glouvermoura@gmail.com
3Mestre em Biotecnologia e Recursos Naturais da Amazônia pela Universidade do Estado do Amazonas (UEA)