REGISTRO DOI: 10.69849/revistaft/cl10202504291703
Brenda Braga da Silva
Bárbara Marques Farias
Gabriela de Avellar Murat
Gabriela Jardim Macedo
Thaynara Cristina Costa Dutra
Thiago Bretz Carvalho
Juliana Eschholz de Araujo*
Abstract
Known as the most common mitochondrial disorder in childhood, Leigh Syndrome is a disease of biochemical and genetic etiology. It specifically affects energy metabolism, being a pathology derived from a genetic alteration. The individual who has this disease presents several clinical manifestations primarily influenced by the phenotypic correspondence to the mutated genes and by the age group, which indicate significant changes in the patient’s prognosis and treatment. The main objective of the research is to present a bibliographical review on Leigh Syndrome, in order to introduce a discussion about this rare disease, clarifying the best examination and treatment methods and highlighting the main symptoms and manifestations that can standardize clinical presentations that come to support the diagnosis of Leigh syndrome. The aim is to contribute to the scientific community’s knowledge and the skills of healthcare professionals. The methodology used consists of a bibliographic review, with a descriptive and integrative character, through documentary research in scientific journals and published articles, cases that can highlight all aspects involving the pathology (etiology, diagnosis and treatments), synthesizing secondary data in qualitative research. The studies confirm the genetic and metabolic bases of the disease, the progression of the disease in early childhood, with involution of motor and cognitive capacity, the main laboratory and imaging diagnostic methods, in addition to genetic screening in the family and treatment, which was proved to be palliative, with extensive use of physiotherapy and vitamin supplementation.
Keywords: Leigh disease, case reports, subacute necrotizing encephalopathy
Introduction
Leigh syndrome (LS) or Leigh’s necrotizing encephalopathy is linked to the group of mitochondrial encephalopathies; it is characterized by a progressive delay and loss of mental and motor abilities, which can lead to death within the first three years of life. LS is caused by a genetic alteration that can be inherited from both the father and the mother, so it is recommended that people with cases of the disease in the family undergo genetic counselling to know the chances of having a child with this syndrome. It is a condition where the modification occurs in energy metabolism, negatively affecting oxidative phosphorylation and cellular ATP production (Roma et al., 2008).
At first glance, the disease in question was described in 1951 by Denis Leigh as a rare condition, with alterations in the energy metabolism of the individual with the syndrome potentially affecting any system, leading, for example, to cardiac and endocrine changes. In this case, the nervous system is the most affected, as it is the system that needs ATP the most. In this way, the individual may exhibit muscle weakness, respiratory problems, and complications in psychomotor development, among other changes (Lopes et al, 2018).
Leigh syndrome can also have harmful multisystem effects on the cardiac, hepatic, gastrointestinal, and renal organs. Biochemical studies in patients with Leigh syndrome tend to show increased lactate and abnormalities in mitochondrial oxidative phosphorylation. (Lake et al, 2016). Moreover, the disease manifests mainly in the first two years of life, with most patients presenting with diseases in the central nervous system (CNS) and the peripheral nervous system (PNS) (Ribeiro, 2015).
The objective of this work was to provide updates on the topic, both for the medical staff, faculty, and healthcare students, as well as for the entire community that needs information about this rare syndrome.
Material and methods
The article consists of an integrative and descriptive literature review that encompasses data from case reports comparatively, drawn from national and international journals and periodicals, as well as from the Pubmed-Medline, Scielo, and Science Direct databases, to characterize fundamental aspects of this disease. The selected case reports were published from 2018 to 2023, in English and Portuguese, totalling 25 articles, which presented 36 cases. Furthermore, along with the case reports, articles and literature review journals, whether recent or not, were selected to outline the fundamental characteristics of the syndrome in the present study from the same databases. The indexed descriptors used in the searches were: “Leigh’s Disease,” “case reports,” and “subacute necrotizing encephalopathy.”
Genetic
Over 75 genes have been associated with Leigh syndrome so far, reflecting the significant genetic heterogeneity of the condition. Similar to other mitochondrial disorders, Leigh syndrome can follow different inheritance patterns, including maternal transmission via mitochondrial DNA mutations and either autosomal recessive or X-linked inheritance when nuclear genes are involved. Even with the growing list of known genes, a considerable number of patients still lack a definitive genetic diagnosis, suggesting that additional causative genes have yet to be discovered (Lake et al., 2015).
Leigh Syndrome presents a wide clinical heterogeneity, mainly due to genetic and biochemical variability. The genetic etiology is confirmed in approximately 50% of cases and identified in nuclear or mitochondrial DNA. Heterogeneous mitochondrial functionality is responsible for the broad clinical manifestations presented in LS due to a dysfunction of a vital area of mitochondrial metabolism, oxidative phosphorylation, where the majority of ATP is produced; this way, any organ can be affected. One of the most frequently mutated mitochondrial genes is the ATPase6 gene (MT-ATP6), which encodes a subunit of complex IV of the respiratory chain, with the most described mutation being the 8993T>G transcript (Lopes et al, 2018).
If LS is caused due to a mutation in nuclear DNA, the disease is inherited either by autosomal recessive inheritance or by X-linked inheritance. Autosomal recessive inheritance means that both parents are healthy carriers of a mutated gene, which means that in each pregnancy of the same parents, the child has a 25% chance of receiving the mutated gene in a double set, a 50% chance of receiving the mutated gene in a single set and a 25% chance of not contracting or becoming a carrier of the disease. The genetic cause of several cases of Leigh syndrome remains unknown, despite the presence of a specific biochemical defect in some cases (Miranda et al, 1989).
Biochemical impairments affecting all five complexes of the oxidative phosphorylation system, as well as the electron carrier coenzyme Q10 (CoQ10), have been identified in patients with Leigh syndrome. Since the pyruvate dehydrogenase complex (PDHc) plays a key role in supplying electron donors to the mitochondrial respiratory chain, its deficiency also disrupts cellular energy production. Among these, complex I deficiency is the most frequently reported abnormality, accounting for nearly one-third of the known genetic causes of the syndrome. While isolated complex IV dysfunction and combined defects involving multiple oxidative phosphorylation complexes are also relatively common, abnormalities in complexes II, III, V, or in CoQ10 biosynthesis occur less often (Lake et al., 2015).
Furthermore, there are mutations involving the T-C transcripts at position 8993 or the A-G transition at position 8344, which cause changes in the pyruvate dehydrogenase complex, as well as the five complexes that make up the respiratory chain, with cytochrome C oxidase deficiency (complex IV) being common in LS (Miranda et al, 1989). Mutations in the nicotinamide adenine dinucleotide (NADH dehydrogenase): ubiquinone Fe-sulfur protein 4 (NDUFS 4) subunit are the most frequent autosomal recessive cause of complex I–associated LS, with > 20 cases reported. And mutations genes SURF 1, encoding a complex IV assembly factor, are the most common cause of complex IV deficient LS and are among the most frequently reported causes of LS, with >200 cases described to date in the literature (Lake et al., 2015).
Clinical presentation
In order to characterize the clinical presentation of Leigh Syndrome (LS) in detail, articles were collected and a total of 36 cases of patients with the disease were studied. All reports share similarities, discrepancies, and characterizations, whether classic or not, of the syndrome under study, and here the main findings will be compiled.
The symptoms most frequently found in patient reports include loss of motor skills (15.9%), cognitive regression (13.3%), ophthalmological symptoms (11.5%), verbal delay (11.5%), seizures (10.6%), hypotonia (9.7%), postural changes (7.1%), behavioral changes (7.1%), respiratory symptoms (6.2%), cardiac symptoms (3.5 %), falls (3.5%) (see graphic, image 1). Generally, intellectual disability and hypotonia follow a first symptom that appears at a young age: the delay or involution of the patient’s psychomotor development, also mainly manifested through speech delay. Increased tone in the lower limbs is also widely described in the literature (Alemao et al., 2022; Than et al., 2021)
As the disease is neurodegenerative, the involution is progressive and can be perceived through poor logical reasoning, difficulties in gait and balance, ataxia, frequent falls, difficulty sitting with stability, inability to raise the head and behavioral disorders, which, in some of these cases, was described as aggressive (Silva et al., 2019)
In one of the reports studied, the boy, 14 years old at the time of the study, was diagnosed, at the age of 7, with a mental age of 3 and had already had language delay since the age of 5 (Vaz et al., 2022). In another case, the change in speech came later: at the age of 7 in the patient who, at the time of the report, was 15 years old (Valenciano et al., 2019). Regardless of the age at which each patient’s developmental regression occurred, it is important to highlight that the first signs of this syndrome generally appear in early childhood.
Most of the cases studied began with the first signs, such as regression in learning and motor skills, which demonstrate how the condition, despite its wide clinical manifestation and possible complications, presents the neurological system as the main affected one, with the majority of its cases referred to and diagnosed by neurologists. Therefore, many of the other symptoms are consequences of such affected neurological function.
Furthermore, seizures, which are very common, can also occur since the first years of life. In some reports, however, only episodes of muscular dystonia have been described.
Such as other complications that could be observed in patients with Leigh Syndrome, the ophthalmological changes may be present in many patients, such as nystagmus, optic atrophy, myopia, strabismus and astigmatism. It is possible that, because of optic atrophy, the patient has a worsening prognosis for vision loss, whether partial or total (Santos et al, 2018).
Regarding respiratory changes, bronchopneumonia is an example of a prognostic complication in some patients (Aretini et al., 2018). When hospitalization becomes necessary, the respiratory component is susceptible to infections, and what is observed is that many patients with this syndrome may die due to respiratory failure. One of the studies describes a boy with LS who was hospitalized after contracting pneumonia, and his condition worsened with the development of respiratory failures that demanded intubation, but despite efforts, he died at the age of 4 years and 1 month (Alston et al., 2020). It is clear, however, that changes in respiratory function such as underlying lung diseases were not evident in the cases.
Among congenital heart diseases, septal defects, ventricular hypertrophy and patent ductus arteriosus have been reported, for example. One of the cases describes, in a 22-year-old boy, endomyocardiopathy and mitochondrial heart disease, being diagnosed with LS based on endomyocardial biopsy, an uncommon method for diagnosing the syndrome (Mauro et al., 2021).
Regarding deformities and other dysmorphic characteristics, the reports give little space for such descriptions, but in some cases, it is possible to identify characteristics despite there being no pattern of dysmorphism for LS. One of the studies describes dysmorphisms apparently present in the patient, a 1-year-old boy at the time of the study: prominent forehead, hypertelorism, wide nasal bridge, anteverted nostrils, flat philtrum, thin upper lips, and high arched palate (Piro et al., 2020).
In addition to these symptoms, other findings were hemiplegia, hemiparesis, autism, motor tremor, scoliosis, genu valgum, severe bruxism and irritability (Alemao et al., 2022; Azevedo, 2019; Silva et al., 2019; Valenciano et al., 2019).
It is also important to mention the importance of identifying signs and symptoms in neonatal patients, so that the disease can be diagnosed early. To identify neonatal LS, a study supported by the National Nature Science Foundation of China highlights: “Neonatal-type LS initially presents with feeding and swallowing disorders, dyspnea, brainstem dysfunction (such as abnormal eye movement behavior and facial muscle weakness) and severe delay in motor development” (Men et al., 2022).
Graph 1. Characterization of the clinical presentation of LS with the most frequently mentioned symptoms (absolute number of articles)

Source. Alemao et al, 2022; Alston et al, 2020; Aretine et al, 2018; Azevedo, 2019; Borna et al, 2020; Ducharlet et al, 2018; Friederich et al, 2020; Gong et al, 2021; Gonzalez-Quintana et al, 2020; Habbane et al, 2020; Kishita et al, 2021;Lopes et al, 2018; Maruo et al, 2021; Men et al, 2022; Oktay et al, 2020; Piro et al, 2020; Saneto et al, 2021; Santos et al, 2018; Silva et al, 2019; Sun et al, 2020; Thanh et al, 2021; Valenciano et al, 2019; Vaz et al, 2019; Veiga et al, 2019; Wen et al, 2022;
General Diagnosis
The diagnosis of the syndrome is based on brain imaging that shows the specific topology of lesions in the brainstem and basal ganglia, often in association with leukodystrophy and cerebral atrophy. Lactate levels are constantly increased in the cerebrospinal fluid and often in the blood. The etiological diagnosis is based on biochemical investigations that look for the underlying defect in energy production. Pyruvate dehydrogenase is tested in leukocytes or skin fibroblast cultures, while oxidative phosphorylation is analysed more completely in muscle or liver (Demczko, 2021)
Prenatal diagnosis may be possible in cases with a known genetic abnormality in a nuclear gene. It is much more difficult when the alteration involves a mitochondrial DNA gene due to heteroplasmy (coexistence of mitochondria with an altered genome and normal mitochondria). When only the biochemical defect is identified, prenatal diagnosis becomes complex due to possible technical difficulties in the biochemical analysis of amniocytes, as well as the possibility that these cells do not express the defect detected in skin fibroblasts (Steffann et al., 2007).
Rahman et al. (1996), describe diagnostic criteria used based on clinical and neuroimaging characteristics, including:
1. Progressive neurological symptoms with motor symptoms and delay in intellectual development;
2. Signs and symptoms of brain stem or basal ganglia disease;
3. Increased lactate levels in blood or brain fluid (CSF);
4. At least one of the following features: (a) features of LS on neuroimaging (dense hyposymmetric lesions on CT scan or hyperintense lesions on T2 MRI) in the basal ganglia; (b) typical post-mortem neuropathological changes; or (c) typical neuropathology in a similarly affected sibling.
Typical regions affected by LS include the putamen, globus pallidus, brainstem, and thalamus, which appear symmetrical and hyperintense on T2 MRI. In the study carried out by Bonfante et al. (2016), basal ganglia lesions were identified in 76% of patients.
Diagnosis in reviewed cases
Among the reviewed cases, cranioencephalic magnetic resonance stands out as one of the methods used for diagnosing LS, making it essential in many reports (Alemão et al., 2022; Men et al., 2022; Vaz et al., 2022).
Genetic counseling was often conducted in cases where the disease was suspected based on the clinical presentation and findings from other tests, aiming to identify which genes might carry the mutation and to determine the heritability of the disease within the family. DNA screening is an extremely important tool for a comprehensive understanding of the disease and assessing the risk of recurrence within the family (Valenciano et al., 2019).
In the cases where mitochondrial gene mutation screening was performed, no pattern was revealed due to the wide heterogeneity of the disease. In one case, a mutation in 8993T>G (MT-ATP6) was identified in a 16-month-old girl (Lopes et al., 2018). In another case, a mutation in m.14453 A>G in the MT-ND6 gene was identified in a 22-year-old boy (Mauro et al., 2021).
Among the laboratory findings, it is important to mention hyperlactacidemia, heteroplasmy on genetic testing, complex IV or complex I deficiency, aminoaciduria, and metabolic acidosis. The main clinical presentations related to these laboratory findings, as well as additional complications, are listed in Table 1. The clinical presentation of the disease is diverse and may include other manifestations or diagnostic approaches.
Table 1. Clinical Presentation and Most Common Characteristics in the Diagnosis of Leigh Syndrome.
First symptoms/ signs | Most common symptoms | Laboratory findings | Complementary findings | Other possible complications |
| Psychomotor delay and regression | Intellectual disability | Heteroplasmy | MRI: on T2= hypersignal in putamen, globus pallidus, brainstem and thalamus | Ophthalmologicalsymptoms |
| Dysarthria | Central hypotonia | Elevated lactate/pyruvate ratio | Genetic tests=Identification of the mutation in mitochondrial or nuclear DNA | Congenital heart disease |
| Seizures | Aminoaciduria | CT= shows demyelination and cortical atrophy | Behavioral disorders | |
| Lower limb hypertonia | Complex IV/I deficiency | MRI= basal ganglia lesions | Postural disorders |
Source. Alemao et al, 2022; Alston et al, 2020; Aretine et al, 2018; Azevedo, 2019; Borna et al, 2020; Ducharlet et al, 2018 ; Friederich et al, 2020; Gong et al, 2021; Gonzalez-Quintana et al, 2020; Habbane et al, 2020; Kishita et al, 2021;Lopes et al, 2018; Maruo et al, 2021; Men et al, 2022; Oktay et al, 2020; Piro et al, 2020; Saneto et al, 2021; Santos et al, 2018; Silva et al, 2019; Sun et al, 2020; Thanh et al, 2021; Valenciano et al, 2019; Vaz et al, 2019; Veiga et al, 2019; Wen et al, 2022;
Prognosis
The prognosis for Leigh Syndrome is involutional and degenerative, with first symptons often appearing in the early years of life, though in some instances, they may emerge later. However, in most cases, the initial signs begin with the child’s difficulty in maintaining logical reasoning, speech and walking, in addition to behavioral changes. Throughout the study, the case reports showed that the age of the patients at the time of the study was more likely to be in early childhood (Graph 2).
Graph 2. Age of patients at the time of the study versus number of reports (absolute number)

Source. Lopes et al, 2018; Santos et al, 2018; Maruo et al, 2021; Azevedo, 2019; Valenciano et al, 2019; Silva et al, 2019; Vaz et al, 2019; Alemao et al, 2022; Veiga et al, 2019; Men et al, 2022; Thanh et al, 2021; Wen et al, 2022; Kishita et al, 2021; Gong et al, 2021; Saneto et al, 2021; Borna et al, 2020; Piro et al, 2020; Habbane et al, 2020; Gonzalez-Quintana et al, 2020; Alston et al, 2020; Friederich et al, 2020; Oktay et al, 2020; Sun et al, 2020; Aretine et al, 2018; Ducharlet et al, 2018
Being of genetic origin, the pattern of heredity was observed in some families. In one of the studies, a 15-year-old girl, when developing speech delay at age 7, raised suspicion of LS because her older brother had already been diagnosed with the Syndrome (Valenciano et al., 2019). In another report, genetic screening showed that the mother had the same mitochondrial mutation as her 16-month-old daughter (Lopes et al., 2018).
Prognostic complications range from the first symptoms appearing in childhood, with the worsening of the condition over the years, in addition to the appearance of new symptoms. It is rare for patients to reach adulthood, and of all the studies collected, only seven managed to live past 18 years of age.
The older patient at the time of the study was a woman who managed to reach the age of 39 (Oktay et al., 2020). She has dysarthria, cerebellar ataxia, reduced fine motor skills of the upper limbs, spastic paraplegia, and weakness of the lower limbs. Her prognosis declined at the age of 4 with worsening gait, and at the age of 27, her walking ability was reduced to less than 10 meters, and she had to drop out of school. However, she is currently able to work in a secure workplace.
The reality for most patients with Leigh syndrome is a difficult prognosis. They begin with psychomotor regression, dysarthria, seizures, frequent falls, difficulty remaining seated, climbing stairs, etc. In newborns, parents often report difficulties during feeding, such as sucking and swallowing.
However, in some of the reports, certain patients managed to improve cognitive abilities, but treatment should not be stopped (Silva et al., 2019).
In emergencies, infections, apnea crises, and seizures are frequent causes of hospitalization. In addition, metabolic acidosis is a common finding in these patients during hospitalization, with elevated serum lactate (Alemao et al., 2022; Men et al., 2022; Veiga et al., 2019).
Treatment
Among the treatments and approaches used for Leigh Syndrome, the study concluded that vitamin supplementation (generally vitamins B1, B6, B2) was the most commonly used (24.4%), followed by physical therapy (22%), the use of Coenzyme Q10 or the synthetic derivative Idebenone (19.5%); anticonvulsants (14.6%) such as topiramate and phenobarbital; medications for psychiatric and behavioral disorders, such as Ritalin, sertraline, benzodiazepines, and Depakote (9.8%); baclofen (7.3%), used as a muscle relaxant; and ORAP (2.4%). Figure 3 shows the distribution of treatments most used by patients in the reports.
Graph 3. Number of citations versus treatments used (absolute number)
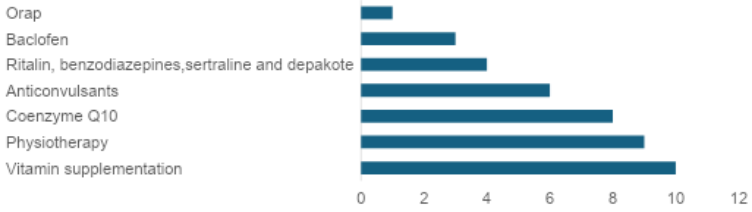
Source. Lopes et al, 2018; Santos et al, 2018; Maruo et al, 2021; Valenciano et al, 2019; Silva et al, 2019; Vaz et al, 2019; Alemao et al, 2022; Men et al, 2022; Thanh et al, 2021; Kishita et al, 2021; Gong et al, 20218; Borna et al, 2020; Friederich et al, 2020; Sun et al, 2020; Aretine et al, 2018.
It is important to emphasize that Leigh Syndrome requires multidisciplinary and palliative treatment, including physical therapy and occupational therapy sessions, to increase the patient’s survival and quality of life.
It is noted that the treatment is solely focused on symptom control, and more advanced measures such as transplants, dialysis, and mechanical ventilation will depend on the prognosis. No specific treatment for Leigh Syndrome or other mitochondrial disorders is found in the literature. However, early diagnosis is essential for the appropriate management to be implemented.
Conclusion
It is noted throughout the literature review that this syndrome has a challenging prognosis, but it can be effectively identified if clinical, laboratory, and imaging aspects are connected for early diagnosis, which has a significant impact on the patients’ quality of life. Since it has a genetic origin, it is important to identify the mutation and conduct family screening. The treatment, however, is palliative and aimed at symptom control.
It is relevant to highlight that due to a lack of adherence to medication or physical therapy, there is often a worsening of the clinical condition, making hospitalization or surgical intervention necessary. Two reports mentioned a worsening of symptoms such as aggressive behavior, loss of motor coordination, poor socialization, and seizures after treatment was discontinued (Silva et al., 2019; Vaz et al., 2022). Therefore, it is important for professionals to instruct caregivers to maintain regular and uninterrupted medication and therapy to enhance the patient’s quality of life and reduce suffering.
Bibliography
Alemao,N.N.G; Gowda,S. Jain, A; Singh,K; Piplani,S.; Shetty, P.D; Dhawan, S; Arya,S; Chugh,Y; Piplani, S. 2022. Leigh’s disease, a fatal finding in the common world: A case report. Radiology Case Reports.V.17, Issue 9. Pages 3321-3325.
Alston C.L.; Veling, M.T.; Heidler, J.; Taylor, L.S.; Alaimo, J.T.; Sung, A.Y.; He, L.; Hopton, S.; Broomfield, A.; Pavaine, J.; Diaz, J.; Leon, E.; Wolf, P.; McFarland, R.; Prokisch, H.; Wortmann, S.B.; Bonnen, P.E.; Wittig, I.; Pagliarini, D.J.; Taylor, R.W. 2020. Pathogenic Bi-allelic Mutations in NDUFAF8 Cause Leigh Syndrome with an Isolated Complex I Deficiency. Am J Hum Genet.;106(1):92-101.
Aretini, P.; Mazzanti, C.M.; La Ferla, M.; Franceschi, S.; Lessi, F.; De Gregorio, V.; Nesti, C.; Valetto, A.; Bertini, V.; Toschi, B.; Battini, R.; Caligo, M.A. 2018. Next generation sequencing technologies for a successful diagnosis in a cold case of Leigh syndrome. BMC Neurol. Jul 2018(1):99.
Azevedo, K.A. 2019. Síndrome de Leigh: Relato de caso Clínico. Universidade Federal de Uberlândia. Faculdade de odontologia.
Bonfante, E.; Koenig, M. K.; Adejumo, R.B.; Perinjelil, V.; Riascos, R.F. 2016. The neuroimaging of Leigh syndrome: case series and review of the literature. Pediatric radiology, 46(4), 443–451. https://doi.org/10.1007/s00247-015-3523-5
Borna, N.N.; Kishita, Y.; Sakai, N.; Hamada, Y.; Kamagata, K.; Kohda, M.; Ohtake, A.; Murayama, K.; Okazaki, Y. 2020. Leigh Syndrome Due to NDUFV1 Mutations Initially Presenting as LBSL. Genes (Basel). Nov 9;11(11):1325.
Dahl, H.H. Chegando ao núcleo dos distúrbios mitocondriais: identificação de genes de enzimas da cadeia respiratória que causam a síndrome de Leigh. (Editorial) Am. J. Hum. Geneta. 63: 1594-1597, 1998. PubMed: 9837811
Demczko, M. Distúrbios do metabolismo do piruvato. Manual MSD. 2021. Mitochondrial Medicine, Children`s Hospital of Philadelphia. Disponível em <https://www.msdmanuals.com/pt-br/casa/problemas-de-sa%C3%BAde-infantil/dist%C3%BArbios-metab%C3%B3licos-heredit%C3%A1rios/dist%C3%BArbios-do-metabolismo-do-piruvato> Acesso em 22 de dez de 2024.
Ducharlet, K.; Thyagarajan, D.; Ierino, F.; McMahon, L. P.; Lee, D. 2018. Perioperative risk assessment for successful kidney transplant in leigh syndrome: a case report. BMC nephrology, 19(1), 23. https://doi.org/10.1186/s12882-018-0816-6
Finsterer, J. 2008. Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol. 39:223-35.
Friederich, M.W.; Elias, A.F.; Kuster, A.; Laugwitz, L.; Larson, A.A.; Landry, A.P.; Ellwood-Digel, L.; Mirsky, D.M.; Dimmock, D.; Haven, J.; Jiang, H.; MacLean, K.N.; Styren, K.; Schoof, J.; Goujon, L.; Lefrancois, T.; Friederich, M.; Coughlin, C.R.; Banerjee, R.; Haack, T.B.; Van Hove, J.L.K. 2020. Pathogenic variants in SQOR encoding sulfide: quinone oxidoreductase are a potentially treatable cause of Leigh disease. 2020. J Inherit Metab Dis.43(5):1024-1036.
Gong, K.; Xie, L.; Wu, Z.S.; Xie, X.; Zhang, X.X.; Chen, J.L. 2021. Clinical exome sequencing reveals a mutation in PDHA1 in Leigh syndrome: A case of a Chinese boy with lethal neuropathy. Mol Genet Genomic Med. Apr;9(4):e1651.
González-Quintana, A.; García-Consuegra, I.; Belanger-Quintana, A.; Serrano-Lorenzo, P.L. A.; Blázquez, A.; Docampo, J.; Ugalde, C.; Morán, M.; Arenas, J.; Martín, M.A. 2020. Novel NDUFA13 Mutations Associated with OXPHOS Deficiency and Leigh Syndrome: A Second Family Report. Genes (Basel). Jul 26;11(8):855.
Habbane, M.; Llobet, L.; Bayona-Bafaluy, M.P.; Bárcena, J.E.; Ceberio, L.; Gómez-Díaz, C.; Gort, L.; Artuch, R.; Montoya, J.; Ruiz-Pesini, E. 2020. Leigh Syndrome in a Pedigree Harboring the m.1555A>G Mutation in the Mitochondrial 12S rRNA. Genes (Basel). Aug 27;11(9):1007
Kishita, Y.; Ishikawa, K.; Nakada, K.; Hayashi, J.I.; Fushimi, T.; Shimura, M.; Kohda, M.; Ohtake, A.; Murayama, K.; Okazaki, Y. 2021. A high mutation load of m.14597A>G in MT-ND6 causes Leigh syndrome. Sci Rep. May 27;11(1):11123.
Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Síndrome de Leigh: um distúrbio, mais de 75 causas monogênicas. 2016. Ana. Neurol. 79: 190-203. PubMed: 26506407
Lopes, T.; Coelho, M.; Bordalo, D.; Bandeira, A.; Vilarinho, L.; Fonseca, P.; Carvalho, S.; Martins, C.; Oliveira, J.G. 2018. Síndrome de leigh: a propósito de um caso clínico com mutação no dna mitocondrial. Revista Paulista De Pediatria, 36(4), 519–523.
Maruo, Y.; Ueda, Y.; Murayama, K.; Takeda, A. 2021. A case report of Leigh syndrome diagnosed by endomyocardial biopsy. Eur Heart J Case Rep.8;5(2)
Men, L.; Feng, J.; Huang, W.; Xu, M.; Zhao, X.; Sun, R.; Xu, J.; Cao, L. 2022. EEE cyanosis as the first symptom of Leigh syndrome associated with mitochondrial complex I deficiency due to a compound heterozygous NDUFS1 mutation: A case report. Medicine (Baltimore). 26;101(34)
Miranda, A.F.; Ishii, S.; DiMauro, S.; Shay, J.W. 1989. Cytochrome C oxidase deficiency in Leigh’s syndrome: genetic evidence for a nuclear DNA-encoded mutation. Neurology;39(5):697-702
Murphy, J.V. Encefalomielopatia necrosante subaguda (doença de Leigh): detecção do estado de portador heterozigoto. 1973. Pediatrics 51: 710-715. PubMed: 4697519
Oktay, Y.; Güngör, S.; Zeltner, L.; Wiethoff, S.; Schöls, L.; Sonmezler, E.; Yilmaz, E.; Munro, B.; Bender, B.; Kernstock, C.; Kaemereit, S.; Liepelt, I.; Töpf, A.; Yis, U.; Laurie, S.; Yaramis, A.; Zuchner, S.; Hiz, S.; Lochmüller, H.; Schüle, R.; Horvath, R. 2020. Confirmation of TACO1 as a Leigh Syndrome Disease Gene in Two Additional Families. J Neuromuscul Dis. 7(3):301-308.
Piro, E.; Serra, G.; Antona, V.; Giuffrè, M.; Giorgio, E.; Sirchia, F.; Schierz, I.A.M.; Brusco, A.; Corsello, G. 2020. Novel LRPPRC compound heterozygous mutation in a child with early-onset Leigh syndrome French-Canadian type: case report of an Italian patient. Ital J Pediatr. Sep 24;46(1):140.
Rahman, S.; Blok, R.B.; Dahl, H.H.; Danks, D. M.; Kirby, D.M.; Chow, C.W., Christodoulou, J., & Thorburn, D. R. (1996). Leigh syndrome: clinical features and biochemical and DNA abnormalities. Annals of neurology, 39(3), 343–351. https://doi.org/10.1002/ana.410390311
Ribeiro, B.S.V.R. 2015. Caracterização clínica, neurorradiológica e genética de pacientes com síndrome de Leigh. Dissertação (Mestrado em Medicina) – Faculdade de Medicina, Universidade Federal de Minas Gerais. Belo Horizonte, p. 108. 2015.
Roma, A. de C., Pereira, P. R. A. de A., & Dantas, A. M. 2008. Síndrome de Leigh: relato de caso. Arquivos Brasileiros De Oftalmologia. 71(1), 118–121.
Saneto, R.P.; Patrick, K.E.; Perez, F.A. 2021. Homoplasmy of the m. 8993 T>G variant in a patient without MRI findings of Leigh syndrome, ataxia or retinal abnormalities. Mitochondrion. Jul;59:58-62.
Santos, A.A; De Mello, F.T; Silva, O.B; Schiavelli, I.A.P; Vagenas, D.D.N.F.A. 2018. A importância do trabalho multiprofissional para Síndrome de Leigh: Relato de Caso The relevance of multiprofessional team for Leigh Syndrome: A Case Report. Revista Saúde e Meio Ambiente, v. 7, n. 3, p. 33-41.
Steffann, J.; Gigarel, N.; Corcos, J.; Bonnière, M.; Encha-Razavi, F.; Sinico, M.; Prevot, S.; Dumez, Y.; Yamgnane, A.; Frydman, R.; Munnich, A.; Bonnefont, J. P. 2007. Stability of the m.8993T->G mtDNA mutation load during human embryofetal development has implications for the feasibility of prenatal diagnosis in NARP syndrome. Journal of medical genetics, 44(10), 664–669. https://doi.org/10.1136/jmg.2006.048553
Silva, B.M; Negreiros,C.B; Teixeira,L.S; Mota, M.S; Oliveira, N.P. T; Silvestre, M. de Andrade. 2019. Síndrome de Leigh: relato de caso. RESU-Revista de Educação em Saúde. V.7. Suplemento 2
Sun, D.; Liu, Z.; Liu, Y.; Wu, M.; Fang, F.; Deng, X.; Liu, Z.; Song, L.; Murayama, K.; Zhang, C.; Zhu, Y. 2020. Novel ECHS1 mutations in Leigh syndrome identified by whole-exome sequencing in five Chinese families: case report. BMC medical genetics, 21(1), 149. https://doi.org/10.1186/s12881-020-01083-1
Thanh Hai, N.; Kim Ngan, V.; Quynh Giang, N.; Huy Hoang, V.; Anh Viet, L.; Truong Duc, N.; Thu Hien, T. T.; He, D. V., Minh Duc, N. 2021. The magnetic resonance imaging of Leigh syndrome in a child. La Clinica terapeutica, 172(6), 500–503. https://doi.org/10.7417/CT.2021.2364
Valenciano, P.; Klhem, A.R.; Pereira, D.M; Tsuzuki, L.; Evaristo, P.; Rodrigues, C.P. 2019. Síndrome de Leigh e implicações para a prática fisioterapêutica: relato de caso. Arch. Health Sci. 26(3): 183-186.
Vaz, M.H.V.; Jaime, A.; Oliveira, A.L.S.; França, P.R.; De Paula, T.O.M, Silvestre, M.A. 2022. Síndrome de Leigh: um relato de caso atípico. RESU-Revista de Educação em Saúde. V.10. Suplemento 1.
Veiga, M.G.A.; Marecos,C.; Duarte, S.T.; Vieira, J.P.; Conceição, C. 2019. Leigh syndrome with atypical cerebellar lesions. E NeurologicalSci. V.16. 100197
Wen, Y.; Lu, G.; Qiao, L.; Li, Y. 2022. A Leigh syndrome caused by compound heterozygous mutations on NDUFAF5 induce early infant death: A case report. Mol Genet Genomic Med. Jan;10(1):e1852
*UNIFAA- University Center of Valença – Medical School of Valença – Rio de Janeiro