REGISTRO DOI: 10.69849/revistaft/ch10202601311255
Carolina Pismel Xavier Pinto
Orientadora: Dra Gabriela Dias Tomaz
RESUMO
O estudo tem como objetivo relatar o caso de Artrogripose Renal Colestática (Síndrome ARC- doença autossômica recessiva rara) de uma paciente de 4 anos acompanhada no serviço de Hepatologia infantil da Santa Casa de Misericórdia do Pará, com revisão integrativa. No estudo, a paciente teve início dos sintomas desde o nascimento, apresentando artrogripose, icterícia colestática neonatal, ictiose, agenesia de corpo caloso, surdez parcial, infecções recorrentes, repercussões nutricionais graves e atraso global no desenvolvimento. O diagnóstico foi desafiador, inicialmente pensado em outras hipóteses diagnósticas, mas com auxílio da história clínica e teste genético, chegou-se à conclusão da patologia. O tratamento incluiu rifampicina, ácido ursodesoxicólico e polivitamínicos. A relevância do estudo está relacionada ao reconhecimento precoce da doença pelos médicos, o que poderá repercutir em tratamento adequado, acompanhamento multidisciplinar e melhoria na qualidade de vida dos afetados.
Palavras- chave: Artrogripose. Colestase. Disfunção renal
ABSTRACT
This study aims to report a case of Arthrogryposis–Renal Dysfunction–Cholestasis Syndrome (ARC Syndrome)—a rare autosomal recessive disorder—in a four-year-old patient followed at the Pediatric Hepatology Service of Santa Casa de Misericórdia do Pará, along with an integrative literature review. The patient presented symptoms from birth, including arthrogryposis, neonatal cholestatic jaundice, ichthyosis, agenesis of the corpus callosum, partial deafness, recurrent infections, severe nutritional repercussions, and global developmental delay. The diagnosis was challenging, as other hypotheses were initially considered; however, with the support of clinical history and genetic testing, the diagnosis of ARC syndrome was confirmed. Treatment included rifampicin, ursodeoxycholic acid, and multivitamins. The relevance of this study lies in the early recognition of the disease by healthcare professionals, which may lead to appropriate treatment, multidisciplinary follow-up, and improved quality of life for affected patients.
Keywords: Arthrogryposis. Cholestasis. Renal dysfunction.
1 INTRODUÇÃO
Diante de um panorama histórico, é possível identificar a primeira descrição da Síndrome de Artrogripose-Disfunção renal-Colestase (ARC) em 1973, reconhecida na prole de um casamento consanguíneo (ZHOU; ZHANG, 2014). Trata-se de uma doença autossômica recessiva rara, que afeta tanto indivíduos do sexo masculino quanto do sexo feminino em proporções iguais, predominante em indivíduos de descendência asiática, causada por mutações no gene VPS33B ou VIPAR (CULLINANE et al., 2010), que estão localizados no cromossomo 15q26.1.
As mutações em VPS33B são detectáveis em aproximadamente 75% dos pacientes com diagnóstico clínico de síndrome ARC (ZHOU; ZHANG, 2014). Tal alteração gera uma biogênese anormal de organelas, visto que o gene VPS33B codifica proteínas essenciais para as vias de tráfego vesicular intracelular, enquanto as proteínas codificadas pelo gene VIPAR exercem efeitos pleiotrópicos na polaridade e distribuição das estruturas proteicas nas membranas celulares, como descrito por Marino et al. (2022). A alteração genética, neste caso, dificulta a geração e manutenção de estruturas teciduais, como ductos biliares e túbulos renais, resultando em colestase e urina anormal. Essas proteínas são encontradas em vários locais do corpo, incluindo os músculos esqueléticos, rins, fígado, pele, coração, pulmões e cérebro, o que explica os sintomas multissistêmicos característicos do fenótipo clínico ARC (CULLINANE et al., 2010).
Tem como manifestações clínicas clássicas a tríade, como o próprio nome já diz: artrogripose, disfunção tubular renal e icterícia colestática neonatal, mas também pode apresentar manifestações secundárias como ictiose e hiperceratose, anormalidades plaquetárias, agenesia de corpo caloso, anomalias cardiovasculares congênitas, surdez, infecções recorrentes, repercussões nutricionais graves e atraso global no desenvolvimento, como descrito por Gissen et al. (2006) e Bermudez, Notejane e Machado (2024)
De acordo com Gupta et al. (2018), a prevalência da síndrome ARC ainda não é bem definida, menos de 100 casos foram relatados na literatura. Geralmente pacientes com esse conjunto de sinais e sintomas clínicos, tem uma expectativa de vida que não ultrapassa um ano na maioria dos casos (TAVEIRA; SAWAMURA, 2021)
A pesquisa de Choi et al. (2022) estima que as dificuldades em reconhecer o quadro clínico levam ao subdiagnóstico. Por isso, distinguir a icterícia causada por colestase de condições não colestáticas (como icterícia fisiológica do recém-nascido, por exemplo) é fundamental, visto que a icterícia por causas colestáticas é provavelmente patológica e, portanto, pacientes com tais alterações clínicas se beneficiariam imediatamente do diagnóstico precoce e da instituição de terapia específica.
O tratamento da síndrome ARC é baseado em medidas de suporte, incluindo hidratação, suplementos nutricionais, tratamento de colestase, prurido e controle de função renal, além de incluir medidas de cuidados paliativos precocemente, visando uma melhor qualidade de vida para a criança e sua família.
2 OBJETIVOS
2.1 Geral
Relatar o caso de uma paciente diagnosticada com a Síndrome Artrogripose–Disfunção renal–Colestase (ARC) e realizar uma revisão de dados bibliográficos já descritos em literatura.
2.2 Específicos
- Identificar os mecanismos fisiopatológicos envolvidos na patogênese da Síndrome ARC;
- Relacionar as manifestações clínicas da doença com as limitações cotidianas e como é possível chegar a um diagnóstico de forma precoce;
- Analisar os tratamentos disponíveis e os que ainda estão em estudo
- Discutir o prognóstico e sobrevida na Síndrome ARC;
- Identificar as falhas de manejo da doença.
3 METODOLOGIA
Inicialmente, foi realizada a coleta de dados a partir de prontuário médico de paciente com diagnóstico de Síndrome ARC e entrevista com responsável pela criança (genitora), com o objetivo de identificar sexo, idade, antecedentes pessoais e familiares, manifestações clínicas, repercussões sistêmicas, associação com outras patologias, diagnóstico e resposta terapêutica.
Posteriormente, foi realizado levantamento bibliográfico referente à Síndrome ARC em base de dados para construção da revisão integrativa, a fim de identificar achados clínico-epidemiológicos entre os portadores da enfermidade. Para a construção desse estudo, foram utilizados artigos indexados no PubMed, ScIELO e Biblioteca Virtual em Saúde dos últimos 10 anos.
Por fim, uma discussão a fim de comparar dados bibliográficos com o caso estudado, na qual busca-se evidenciar as concordâncias e divergências entre ambos.
Tipo de Estudo
Trata-se de um relato de caso clínico de natureza descritiva, com abordagem qualitativa, realizado a partir da análise documental e observacional do atendimento prestado.
Local do Estudo
O estudo foi realizado no ambulatório de Hepatologia Infantil da Fundação Santa Casa de Misericórdia do Pará, em Belém-Pará, onde a paciente mantém acompanhamento ambulatorial.
Critérios de inclusão
Neste relato de caso foi incluída uma paciente que atende aos seguintes critérios: diagnóstico confirmado de Síndrome ARC, de acordo com os critérios clínicos, laboratoriais e genéticos estabelecidos em literatura; disponibilidade de informações clínicas e exames complementares necessários para análise do caso; consentimento livre e esclarecido assinado pela responsável legal (paciente menor de idade), autorizando o uso das informações clínicas para fins acadêmicos e científicos.
Critérios de Exclusão
Foram excluídos da análise: casos com diagnóstico clínico ou laboratorial inconclusivo de Síndrome ARC e falta de informações clínicas relevantes ou prontuário incompleto.
Coleta de Dados
A coleta de dados foi realizada por meio da revisão do prontuário eletrônico da paciente, complementada pela análise de teste genético, exames laboratoriais e de imagem, arquivos pessoais da família e registros fotográficos do quadro clínico (obtidos pela câmera do celular da própria genitora), quando disponíveis. Também foram consideradas informações obtidas durante o acompanhamento ambulatorial do paciente. Foram utilizados dados do período de Janeiro de 2023 até Setembro de 2025.
Aspectos Éticos
O estudo seguiu as diretrizes da Resolução 466/2012 do Conselho Nacional de Saúde (CNS) bem como a resolução 510/2016 e 580/2018, garantindo sigilo e confidencialidade das informações coletadas. O projeto foi submetido ao Comitê de Ética em Pesquisa (CEP) da Fundação Santa Casa de Misericórdia do Pará, sendo aprovado para publicação conforme parecer consubstanciado nº 7.818.976 . O TCUD- Termo de Compromisso e Utilização de Dados, o TCLE – Termo de Consentimento Livre e Esclarecido, o Termo de Assentimento Livre e Esclarecido- TALE e a Declaração para uso de imagem, voz e relatos escritos foram assinados por todos os pesquisadores envolvidos na coleta, conforme a Norma Operacional da CNS 001/2013. O responsável legal pela paciente foi devidamente informado sobre os objetivos e natureza da publicação e também assinou os termos (TCLE, o TALE e a Declaração para uso de imagem e voz), autorizando o uso de seus dados clínicos de forma anônima.
4 RELATO DE CASO
Paciente do sexo feminino, nascida de 42 semanas, por parto cesáreo de emergência devido sinais de sofrimento fetal agudo (taquicardia fetal) em um hospital particular de Belém-PA, com Apgar 8/8, pesando 3.835 gramas. A gravidez foi planejada e a genitora já havia tentado engravidar previamente por 4 anos consecutivos, sem sucesso (devido diagnóstico de Síndrome do Ovário Policístico). Única gestação, nenhum aborto. Durante o pré-natal, a mãe evoluiu com pré-eclâmpsia, diabetes gestacional e colelitíase. Os pais não são consanguíneos. Ao nascimento, evoluiu com hipoglicemia, icterícia e hipoatividade no primeiro dia de vida. Apresentava dificuldade de sucção ao seio materno, recebendo fórmula infantil de partida com auxílio de copinho, foi colocada sob fototerapia e permaneceu internada na unidade neonatal por 2 dias. Após melhora, recebeu alta médica.
Em torno de 5-6 meses de vida, começou a ter vômitos em jato associado a distensão abdominal e atraso no desenvolvimento neuropsicomotor ao longo de todo primeiro ano de vida (sentou com apoio aos 8 meses, andou com 3 anos). Durante o processo de introdução alimentar, que foi realizado somente com 1 ano de idade, a criança apresentava disfagia associada a engasgos. Inicialmente, a genitora não aceitava que poderia haver algo de errado com a filha, mas ao completar 1 ano e 1 mês, a mesma começou a apresentar sonolência excessiva, astenia, febre, acolia e prurido intenso, motivos pelos quais foram em busca de atendimento médico. Desde então, fez acompanhamento com médico hepatologista de forma ambulatorial, que interrogou dentre os diagnósticos diferenciais, Doença de Wilson e Colestase Intra-Hepática Familiar Progressiva (PFIC).
Logo em seguida, foi internada devido piora clínica, evoluiu com distúrbios hidroeletrolíticos e ácido-básicos, distensão abdominal, colúria, piora da icterícia, desidratação e estava com baixo peso para idade, em grau de magreza acentuada (figura 1). Os testes de função hepática indicaram hiperbilirrubinemia direta com elevação das enzimas hepáticas, mantendo níveis normais de gama glutamil transpeptidade (GGT). Outras causas de icterícia colestática, como infecções virais, também foram descartadas por sorologias. A média dos resultados dos exames laboratoriais por ano e também os resultados dos principais exames de investigação realizados estão dispostos nas tabelas 1 e 2, respectivamente.
Figura 1- Aspectos clínicos apresentados pela paciente
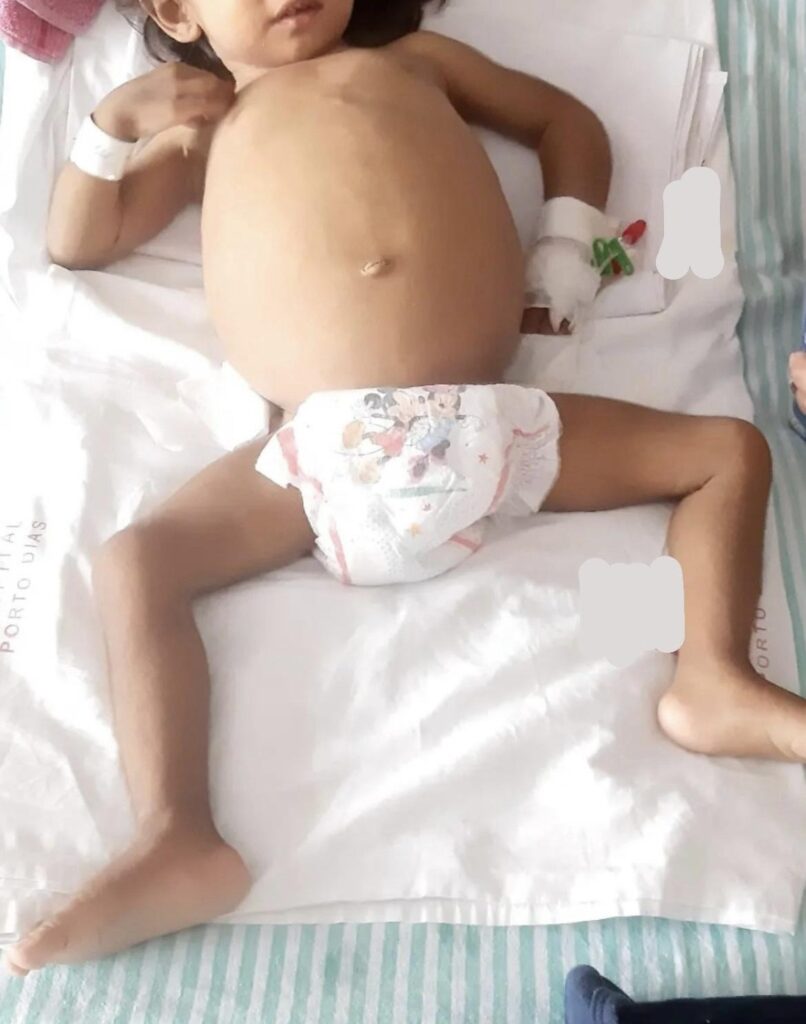
Fonte: Cedida pela mãe da paciente, mediante consentimento informado (2023)
Tabela 1- Média dos resultados de exames laboratoriais por ano.
| ANO | TGO (U/L)[16-57] | TGP (U/L)[19-59] | BT (mg/dL)[até 1,2] | BD (mg/dL)[até 0,4] | BI (mg/dL)[até 0,8] | GGT (U/L)[7-35] | FA (U/L)[73-390] |
| 2023 | 145 | 104 | 14,9 | 10,3 | 4,7 | 30,2 | |
| 2024 | 517 | 499 | 4 | 2,69 | 1,38 | 23,7 | 708 |
| 2025 | 331 | 385 | 0,38 | 0,19 | 0,19 | 21,1 | 982 |
Fonte: registro de prontuário médico, acessado em 2025.
Tabela 2– Exames de imagem, teste genético, cariótipo e biópsia..
| Ressonância Magnética de Crânio (realizada em 05/04/2023) | Leve ectasia com deformidade dos ventrículos laterais e digenesia do corpo caloso. |
| Colangioressonância (realizada em 21/06/2023) | Vesícula normodistendida, com paredes finas e conteúdo homogêneo; Ausência de dilatação das vias biliares intra e extra-hepáticas. Colédoco de calibre normal, sem cálculos e volume normal; Ducto pancreático normal; Fígado discretamente aumentado de volume, com superfície preservada. |
| Biópsia de fígado (realizada em 28/08/2023) | Estrutura lobular preservada e um total de doze espaços porta sem inflamação e com ductos biliares e vasos sem alterações. Não se observa proliferação ductular biliar ou ectasia periportal. Os hepatócitos apresentam tumefação turva focalmente. Os sinusóides estão dilatados. Não se observa inflamação lobular, alteração regenerativa na forma de pseudoroseta, colestase, colestase intracanalicular ou intrahepatocítica, necrose perivenular, granuloma ou outras alterações morfológicas. |
| Cariótipo com Banda G (realizado em 13/03/2023) | Número de células analisadas: 20 Resolução: 400 bandas Cariótipo: 46, XX |
| Teste Genético- Painel de Colestase Crônica (realizado em 03/08/2023) | Síndrome artrogripose-disfunção renal- colestase tipo 1 (gene VPS33B) |
| Ultrassonografia de rins e vias urinárias (realizado em 16/06/2023) | Rins tópicos, de forma, contornos e dimensões habituais. A espessura e ecogenicidade do parênquima renal estão preservados. Rim direito mede: 6,6×3,2×3,4 cm Rim esquerdo mede: 6,7×2,6×3,2 cm Sem evidências de anormalidade. |
Fonte: registro de prontuário médico, acessado em 2025
Ainda durante a internação, permaneceu recebendo dieta por sonda nasoenteral e apresentou refluxo ácido-patológico em exame de phmetria, evoluindo com indicação de confecção de gastrostomia. Também apresentava hepatomegalia de 5 centímetros. Após estabilização clínica, a paciente foi submetida a laparotomia exploradora e biópsia hepática (resultado na tabela 3). Além das alterações já descritas, a paciente também tem hipertricose mais acentuada nos membros superiores, mancha hipercrômica na região sacral, dismorfias faciais (região frontal estreita, narinas antevertidas e filtro curto) e testes auditivos que identificaram perda auditiva moderada bilateralmente, por isso foi encaminhada para avaliação com geneticista, onde foi solicitado o exame CGH-array, um exame genético molecular que analisa o número de cópias de sequências de DNA, detectando perdas (deleções) ou ganhos (duplicações) de material genético. É mais sensível que o cariótipo para identificar alterações cromossômicas não reconhecíveis clinicamente, porém o exame não foi realizado devido ao alto custo.
Atualmente, a paciente realiza acompanhamento multidisciplinar com equipe médica, fonoaudiologia, fisioterapia e nutrição. Faz uso de rifampicina para controle de prurido, com êxito parcial. Anteriormente também já fez uso de fenobarbital, mas sem melhora. Está participando de um estudo denominado “Estudo com uso de Maralixibat em pacientes com colestase e prurido (não alagille/ não PFIC/ não colestase extra-hepática)”, porém ainda não eleita para uso da medicação devido persistência de alteração das enzimas hepáticas acima do ponto de corte. Recebe também ácido ursodesoxicólico, suplementação de vitaminas lipossolúveis e laxativo osmótico para evitar encefalopatia hepática. Paciente sem indicação de transplante hepático até o momento, mas sendo submetida a nova avaliação para investigar se houve mudança no anatomo-patológico.
5 DISCUSSÃO
A colestase neonatal (CN) é definida como diminuição da formação ou fluxo biliar devido a disfunção hepatobiliar ou colangiopatia obstrutiva no período neonatal. A principal falha é a excreção de bilirrubina, resultando em excesso de bilirrubina direta no sangue e diminuição de sais biliares no trato gastrointestinal. A colestase (icterícia) pode ser resultado de disfunção intra-hepática ou extra-hepática, embora algumas condições possam se sobrepor (CHOI HJ, et al., 2022).
O quadro clínico geralmente se apresenta com icterícia, acolia, prurido, colúria, hepatoesplenomegalia, atraso global no neurodesenvolvimento e, em alguns casos, malformações congênitas, como polisplenia/asplenia, doenças cardíacas congênitas, má rotação do intestino e anomalias esqueléticas (BILAL H, et al., 2022). A má absorção de gorduras e vitaminas lipossolúveis (A, D, E e K) devido à bile insuficiente no trato gastrointestinal leva a deficiências de vitaminas, deficiências nutricionais e baixo crescimento (YANG X, et al., 2022)
Bioquimicamente, a CN é caracterizada por níveis séricos elevados de bilirrubina direta, transaminases e fosfatase alcalina. A Gama Glutamiltransferase (GGT) pode ser normal ou baixa (Fawaz R, et al.2017). A icterícia colestática é caracterizada por hiperbilirrubinemia direta (quando a direta é maior que 20% da bilirrubina total (ou > 1 mg/dL, se bilirrubina total < 5 mg/dL) (FISCHER HS, et al., 2020). Outros exames também devem ser solicitados para investigação da provável etiologia, como hemograma, incluindo reticulócitos e contagem de plaquetas, eletrólitos, ureia, creatinina, coagulograma, TGO, TGP, fosfatase alcalina, GGT, proteínas totais e frações, glicemia, TSH, T4L e sorologias.
Como o diagnóstico diferencial é amplo, os exames de imagem devem ser solicitados conforme a principal suspeita, direcionada pelos achados clínicos em conjunto com os resultados de exames laboratoriais já obtidos. Existem diferentes níveis de investigação no lactente colestático, enfatizando a necessidade de individualizar cada caso, a idade da criança e o estágio da doença. A ultrassonografia abdominal geralmente é o primeiro exame devido à alta disponibilidade; não é invasiva e pode avaliar o tamanho do fígado e certas anormalidades na vesícula biliar e no ducto biliar comum. No entanto, não é específico. A cintilografia de vias biliares (com radiofármaco DISIDA) é utilizada para avaliar obstrução das vias biliares extra-hepáticas (RASHED YK, et al., 2013). Segundo os estudos de Boskovic A, et al. (2014), quando um diagnóstico não é feito, a biópsia hepática e, às vezes, a colangiografia cirúrgica geralmente são feitas precocemente, bem como avaliação de painel de genes/exoma.
As causas intrahepáticas podem ser infecciosas, aloimunes, metabólicas/genéticas ou tóxicas. As infecções podem ser de etiologias virais (herpes simples, citomegalovírus, rubéola), bacterianas (bactéria gram-positiva e gram-negativa) ou parasitárias (toxoplasmose) (WANG NL, et al., 2020). Entre as causas metabólicas, existem muitos erros inatos do metabolismo, como galactosemia, tirosinemia, deficiência de alfa-1-antitripsina, distúrbios do metabolismo lipídico, deficiências de ácidos biliares, doenças mitocondriais e deficiências de oxidação de ácidos graxos. Outros defeitos genéticos incluem síndrome de Alagille, fibrose cística e síndrome de artrogripose-disfunção renal-colestase (ARC). Existem também mutações genéticas que interferem na produção e excreção normais da bile; as doenças resultantes são chamadas colestase intra-hepática progressiva familiar (PFIC) (Feldman AG, Sokol RJ. 2019).
A colestase é apenas um sintoma, que deve ser tratado de acordo com a sua causa. Para lactentes com suspeita de atresia das vias biliares (AB), por exemplo, é necessário exploração cirúrgica com colangiografia intraoperatória. (Harpavat S, et al. 2020). O prurido (ictiose) é decorrente da colestase e pode surgir tanto na doença aguda quanto na crônica de causa intra ou extra-hepática, ocorrendo em 20% a 50% dos pacientes ictéricos. Pode ser localizado ou generalizado, contínuo ou intermitente. É, às vezes, relatado como uma sensação de queimação, formigamento ou de um inseto caminhando sobre a pele. Inicia-se, em geral, na palma das mãos e planta dos pés, e progride para superfície extensora dos membros superiores, face, ouvido e região superior do tronco. A sua intensidade também é variável, podendo ser leve, moderada ou intensa (AZEVEDO RA, et al. 2002).
O tratamento do prurido da colestase deve se iniciar por medidas gerais, sendo fundamental a hidratação da pele para evitar o seu ressecamento. Além disso, também existem algumas opções medicamentosas que visam a diminuição da quantidade de substâncias ditas pruritogênicas no organismo. Como exemplo, a colestiramina (ligando-se aos ácidos biliares e a outros componentes orgânicos, impedindo, assim, a absorção e promovendo sua excreção fecal); a rifampicina e o fenobarbital (drogas que ativam o sistema oxidativo microssomal do fígado, levando a um possível aumento da metabolização e excreção de substâncias pruritogênicas endógenas). A rifampicina, por sua vez, parece ser mais eficaz em aliviar o prurido e induzir melhora laboratorial, principalmente da GGT, em particular nas crianças. O mecanismo responsável parece ser a ativação do citocromo P450 monooxigenase, que promove a hidroxilação dos ácidos biliares e estimula sua excreção renal (AZEVEDO RA, et al. 2002).
Agentes mais novos sob consideração para o tratamento do prurido colestático incluem inibidores do transportador do ácido biliar ileal (IBAT) e inibidores de autotaxina. O IBAT representa um alvo terapêutico interessante para a interrupção do ciclo entero‐hepático, aumentando assim a secreção de sais biliares pelo trato intestinal. São exemplos de inibidores do IBAT: linerixibat, maralixibat e odevixibat. Outro medicamento para o controle do prurido é o agonista do receptor κ‐opioide, nalfurafina, aprovado no Japão para o tratamento do prurido colestático desde 2015 (Nietsche TR, et al., 2022)
Uma das causas, extremamente rara, de colestase neonatal é a síndrome ARC, que é uma doença multissistêmica fatal que afeta principalmente o fígado, os rins, a pele, os sistemas nervoso central e musculoesquelético. Tem caráter progressivo e herança autossômica recessiva. Neste estudo, foi apresentado o caso de uma paciente do sexo feminino que começou a manifestar os primeiros sintomas desde o nascimento, porém só percebido pela genitora, de fato, com 1 ano de idade, sendo diagnosticada 6 meses após o início das investigações clínicas. A paciente apresentava sintomas clínicos e teste genético confirmatório para Síndrome ARC, o que corroborou para o diagnóstico. O curso clínico da doença teve uma série de complicações, principalmente associado a distúrbios metabólicos.
Em literaturas mais antigas, o óbito ocorria antes dos 9 meses de vida, com exceção de um caso, relatado por Coleman et al. (1997), que sobreviveu até os três anos de idade, apresentando cirrose e retardo no desenvolvimento psicomotor (MELEAN GG, et al. 2007). Até o ano de 2014, existiam 49 mutações patogênicas do gene VPS33B e 14 mutações patogênicas de VIPAR listadas no Banco de Dados de Variações de Código Aberto de Leiden (LOVD) para ARC, de acordo com Zhou Y. e Zhang J. em 2014.
Apesar de que a expectativa de vida estimada na maioria dos estudos da base de dados ainda é, em média, até os 2 anos de idade, existem estudos como o de Zhu Y. e Chen D. em 2022, relatando mutações inéditas no gene VPS33B de 19 pacientes, que tiveram uma sobrevida prolongada (acima dos 2 anos de idade), sendo uma delas com 12 anos de idade. A criança relatada neste trabalho tem, atualmente, 4 anos. Na pesquisa de YU. et al (2022) também foi abordado sobre fenótipos causados por mutações em VPS33B, demonstrando que algumas variações podem ser mais leves, não apresentando todas as manifestações, consequentemente, levando o portador da síndrome a ter uma vida mais longa.
De acordo com Mutlu M. et al (2010), a artrogripose é uma característica fundamental da síndrome ARC e se manifesta como desvio radial do punho, luxação da articulação, tálus vertical, pé plano, fratura patológica, cifose rígida, pé calcâneo-valgo, atrofia muscular e contraturas em flexão dos membros. No caso da paciente, estavam presentes pé plano, atrofia muscular, contratura em flexão dos cotovelos e displasia de quadril.
A disfunção tubular renal é outra característica da síndrome ARC. Caracteriza-se por acidose tubular renal, diabetes insípido nefrogênico, glicosúria, aminoacidúria, fosfatúria e distúrbios hidroeletrolíticos. No caso da paciente, estavam presentes glicosúria, proteinúria, acidose metabólica e hiponatremia.
A icterícia colestática neonatal é o que completa a tríade da síndrome ARC. Hepatomegalia, icterícia colestática com GGT consistentemente baixo, é a característica mais comum da doença. Entretanto, também existem casos com altos níveis de GGT causada pela mutação VPS33B (Cullinane AR, et al. 2010) Os níveis de AST e ALT podem ser normais ou ligeiramente elevados. No caso da paciente, apresentava hepatomegalia, icterícia colestática com níveis baixos de GGT e altos de AST e ALT.
Doenças cardíacas congênitas e anomalias do sistema nervoso central podem ocasionalmente ser observadas em pacientes com síndrome ARC (Zhou Y, Zhang J. 2014). Nossa paciente apresenta agenesia de corpo caloso, evoluindo com atraso do desenvolvimento neuropsicomotor. Outras características adicionais da síndrome ARC, incluindo ictiose, infecções de repetição e desnutrição energético-proteica também estavam presentes e que inclusive, leva a internações hospitalares frequentes.
6 CONCLUSÃO
O diagnóstico precoce e as medidas terapêuticas são essenciais para otimizar os resultados e melhorar a qualidade de vida dos pacientes com síndrome ARC. O prognóstico depende do tipo de mutação genética, do fenótipo e do início da doença, com mecanismos ainda não totalmente compreendidos. Em fenótipos mais leves, a sobrevida pode se estender até a idade adulta precoce. Apesar das pesquisas, a doença é incurável, e a maioria das crianças morre antes do primeiro ano de vida devido a infecções recorrentes e hemorragias internas.
REFERÊNCIAS
- BERMUDEZ, Amanda; NOTEJANE, Martín; MACHADO, Karina. Síndrome artrogriposis – disfunción renal – colestasis. Abordaje diagnóstico y terapéutico del primer caso reportado en Uruguay. Archivos de Pediatría del Uruguay, Montevideo, v. 95, n. 2, e318, 2024. DOI: https://doi.org/10.31134/ap.95.2.17. Disponível em: http://www.scielo.edu.uy/scielo.php?script=sci_arttext&pid=S1688-12492024000301318&lng=es&nrm=iso. Acesso em: 19 fev. 2025.
- CHOI, H. J. et al. Clinical characteristics of neonatal cholestasis in a tertiary hospital and the development of a novel prediction model for mortality. EBioMedicine, v. 77, p. 103890, 2022. DOI: https://doi.org/10.1016/j.ebiom.2022.103890.
- CULLINANE, A. R. et al. Mutações em VIPAR causam artrogripose, disfunção renal e fenótipo de síndrome de colestase com defeitos na polarização epitelial. Nature Genetics, v. 42, n. 4, p. 303-312, 2010. DOI: https://doi.org/10.1038/ng.538.
- GISSEN, P. et al. Características clínicas e genéticas moleculares da síndrome ARC. Human Genetics, v. 120, n. 3, p. 396-409, 2006. DOI: 10.1007/s00439-006-0232-z.
- GUPTA, V. et al. Síndrome de artrogripose, disfunção renal e colestase (ARC): uma associação rara com alto nível de GGT e rim ausente. BMJ Case Reports, v. 2018, p. bcr2017223715, 2018. DOI: 10.1136/bcr-2017-223715.
- MARINO, B. C.; SANDY, N. S.; HESSEL, G.; BRANDÃO, M. A. B. Artrogripose renal colestática diagnosticada a partir de colestase neonatal: relato de caso. Residência Pediátrica, v. 12, n. 3, p. 1-4, 2022. DOI: 10.25060/residpediatr-2022.v12n3-352.
- Nietsche TR, Dotta G, Barcaui CB, Ferraz MLCG. Cholestatic pruritus: a knowledge update. An Bras Dermatol. 2022;97:332–7.
- TAVEIRA, Adriana Távora de Albuquerque; SAWAMURA, Regina. Colestase na infância. In: TRATADO DE PEDIATRIA. 5. ed. São Paulo: Manole, 2021. v. 1, p. 3729.
- ZHOU, Y.; ZHANG, J. Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome: from molecular genetics to clinical features. Italian Journal of Pediatrics, [S.l.], v. 40, p. 77, 20 set. 2014. DOI: https://doi.org/10.1186/s13052-014-0077-3. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4422138/. Acesso em: 29 mar. 2025.
- Zhu Y, Chen D. Two novel mutations in VPS33B gene cause a milder ARC syndrome with prolonged survival in a 12-year-old patient: Case report. Front Pediatr. 2022 Dec 7;10:1041080. doi: 10.3389/fped.2022.1041080. PMID: 36568436; PMCID: PMC9768213.
- YU, L.; LI, D.; ZHANG, T.; XIAO, Y.; WANG, Y.; GE, T. One case of arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome featuring an incomplete and mild phenotype. BMC Nephrology, v. 23, n. 1, p. 228, 27 jun. 2022. DOI: [10.1186/s12882-022-02851-2](https://doi.org/10.1186/s12882-022-02851-2).
- SAAD, A.; CHAUHAN, A.; TRIPATHI, S.; KUMAR, M.Arthrogryposis, renal dysfunction, cholestasis syndrome in a neonate: an uncommon association of common problems. BMJ Case Reports, v. 16, n. 5, p. e254822, 18 maio 2023. doi: 10.1136/bcr-2023-254822.